Article Text

Abstract
Background: Weight loss, mostly due to skeletal muscle atrophy, is a frequent and clinically relevant problem in patients with chronic obstructive pulmonary disease (COPD). The molecular mechanisms underlying this phenomenon are unclear. This study sought to investigate whether activation of the nuclear transcription factor NF-κB and upregulation of the inducible form of nitric oxide synthase (iNOS) occur in the skeletal muscle of patients with COPD and low body weight as potential molecular mechanisms leading to cachexia
Methods: NF-κB DNA binding activity was determined by electromobility shift assay and the immunoreactivity of its inhibitory subunit IκB-κ and that of iNOS by Western blot analysis in biopsy specimens of the quadriceps femoris muscle of seven COPD patients with normal body mass index (BMI, 27.5 (1) kg/m2) and seven patients with low BMI (18.5 (1) kg/m2).
Results: Compared with patients with normal body weight, those with low BMI showed a 30% increase in NF-κB DNA binding activity, a lower expression of IκB-α (3.37 (0.47) IOD v 5.96 (0.75) IOD, p<0.05; mean difference 2.59; 95% CI −4.53 to −0.65) and higher iNOS expression (1.51 (0.29) IOD v 0.78 (0.11) IOD, p<0.05; mean difference 0.74; 95% CI 0.04 to 1.42).
Conclusions: NF-κB activation and iNOS induction occur in skeletal muscle of COPD patients with low body weight. These changes might contribute to the molecular pathogenesis of cachexia in COPD.
- cachexia
- chronic obstructive pulmonary disease
- nitric oxide
- weight loss
Statistics from Altmetric.com
Patients with chronic obstructive pulmonary disease (COPD) often lose weight during the course of their disease.1 This has important clinical implications because it worsens their prognosis2 and quality of life.3 It is therefore important to develop rational and effective new treatments to palliate weight loss in COPD.4 However, because its pathogenesis is unclear,5,6 a better understanding of the underlying mechanisms is essential.4
Atrophy of skeletal muscle is the main cause of weight loss in COPD.4,7 The precise cellular and molecular mechanisms leading to skeletal muscle atrophy in these patients are unclear.5,6 Inactivity, systemic inflammation, oxidative stress, tissue hypoxia, and enhanced skeletal muscle apoptosis have been considered, among others, to be potential pathogenic factors.8 Because systemic inflammation9 and hypoxia10 are particularly prevalent among COPD patients with low body weight and both are well known inducers of the inducible isoform of the nitric oxide synthase (iNOS) through the nuclear transcription factor NF-κB pathway,11,12 we hypothesised that activation of NF-κB and upregulation of iNOS may occur in the skeletal muscle of COPD patients with low body weight. This may constitute a molecular mechanism leading to cachexia in these patients because iNOS upregulation can cause protein nitrotyrosination and favour protein degradation through the ubiquitin-proteasome pathway13 and enhance skeletal muscle apoptosis,14 an event which occurs in COPD patients with low body weight.15
To investigate this hypothesis we compared (1) the degree of NF-κB DNA binding activity by electrophoretic mobility shift assay (EMSA) and the immunoreactivity of its inhibitory subunit (IκB-α) by Western blot analysis, and (2) the immunoreactivity of iNOS by Western blot analysis in biopsy specimens from the quadriceps femoris muscle obtained from COPD patients with normal (n = 7) or low (n = 7) body weight.
METHODS
Patients
The diagnosis of COPD was established according to the British Thoracic Society guidelines.16 The patients were always studied under conditions of clinical stability, which were defined by the absence of treatment change and/or the need for acute medical care during the previous 2 months. All patients received inhaled bronchodilator treatment. Nine patients also received treatment with inhaled steroids, but none had received oral steroids. Low body weight was defined as a body mass index (BMI) of <21 kg/m2.15
The ethical review board of our institution approved the study and all participants signed the informed consent from.
Lung function
Forced spirometric parameters (GS, Warren E Collins, Braintree, MA, USA) and arterial blood gas tensions (IL BG3, Izasa, Spain) were measured in all patients (table 1). Spirometric reference values were those of a Mediterranean population.17
Mean (SE) clinical and functional data of COPD patients included in the study
Muscle biopsy
A percutaneous needle biopsy was obtained from the lateral portion of the quadriceps femoris muscle (at the mid thigh level) under local anaesthesia, as previously described.15,18 Muscle samples were immediately frozen in liquid nitrogen and stored at −80°C until analysis.
Immunodetection of iNOS, IκB-α, and α-tubulin
Immunoblots were performed as previously described in our laboratory.12 In brief, frozen muscle samples (5–10 mg) were homogenised (1:10 w/v) in cold 10 mM Tris buffer pH 7.5 containing 1% sodium dodecyl sulphate (SDS) and a cocktail of protease inhibitors (Roche Diagnostics, Mannheim, Germany). Samples were centrifuged at 12 000 g for 10 minutes at 4°C. The resulting supernatant was collected and proteins were determined by the method of Bradford.19 One hundred μl of the resulting supernatant was mixed with an equal volume of loading buffer (125 mM Tris, pH 6.8, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol and 0.005% bromophenol blue) which was then boiled for 4 minutes.19 Five to 20 μl of the resulting suspension were loaded in a 10% polyacrylamide gel and submitted to SDS-polyacrylamide gel electrophoresis. Proteins were transferred to nitrocellulose membranes (immunoblot, Western blot) incubated in phosphate buffered saline (PBS) containing 4% non-fat dry milk (blocking solution) for 1 hour at room temperature with gentle rocking. The membranes were first incubated over night at 4°C in blocking solution containing the primary antibodies and then, after washing, with the secondary antibodies. The following primary antibodies were used: anti-iNOS rabbit polyclonal antibody at 1:100 dilution (Transduction Laboratories, San Diego, CA, USA), anti-IκB rabbit polyclonal antibody at 1:1000 dilution (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA), and anti-α-tubulin mouse monoclonal antibody (Sigma Chemical Co, St Louis, MO, USA) at 1:2000 dilution. The secondary antibodies, a horseradish peroxidase linked sheep anti-rabbit IgG for iNOS and IκB-α and a horseradish peroxidase linked donkey anti-mouse IgG for α-tubulin (Amersham International, Buckinghamshire, UK) were both incubated at 1:1000 dilution in blocking solution at room temperature for 2 hours. The specificity of the anti-iNOS antibody was determined by Western blot analysis of a positive control of purified iNOS from mouse macrophages (Transduction Laboratories) and by positive controls performed in our laboratory by inducing iNOS expression in A549 cells with a mixture of cytokines and lipopolysaccharide as described by Asano et al.20 Immunoreactivity was detected with a chemiluminescence Western blot detection system (Pierce, Rockford, IL, USA) with the aid of a chemiluminescence sensitive video camera system (Syngene, Cambridge, UK). The results were normalised to the α-tubulin content.
Electrophoretic mobility shift assay (EMSA)
Because of the relatively low yield of protein in needle biopsies of skeletal muscle, NF-κB activation was determined in pooled nuclear extracts as other investigators have done previously.21 Nuclear proteins (20 μg) were extracted from muscle samples by detergent lysis22 and incubated for 20 minutes in binding buffer (10 mM Tris, 50 mM KCl, 5 mM MgCl2, 1 mM DTT at pH 7.5) containing 2.5% glycerol, 1% NP-40 with 20 fmol biotin labelled double stranded oligonucleotide containing the consensus sequence for the NF-κB-DNA binding site (5′-AGTTGAGG GGACTTTCCCAGG-3′). Specificity was determined by the addition of a 200-fold excess of unlabelled double stranded NF-κB. DNA-protein complexes were resolved on a 6% non-denaturing polyacrylamide gel in 1X Tris-borate-EDTA buffer. The retarded bands were detected by chemiluminescence using the LightShift Chemiluminescent EMSA kit (Pierce). To confirm the identity of NF-κB binding, supershift experiments were performed using anti-p65 human antibody (Santa Cruz Biotechnology Inc). Briefly, EMSA gels were transferred to nitrocellulose membranes that were incubated in blocking solution for 1 hour at room temperature with gentle rocking. The membranes were then incubated over night at 4°C in blocking solution containing anti-NF-κB p65 primary rabbit polyclonal antibody at 1:1000 dilution (Santa Cruz Biotechnology). The secondary antibody, a horseradish peroxidase linked sheep anti-rabbit IgG (Amersham International) was incubated at 1:1000 dilution in blocking solution at room temperature for 2 hours. The retarded bands (EMSA) and immunoreactivity were detected with a chemiluminescence detection system (Pierce) with the aid of a chemiluminescence sensitive video camera system (Syngene). The bands were counted by densitomery with the aid of Genesnap and Genetools software (Synoptics Ltd, Cambridge, UK).
Statistical analysis
The results are presented as mean (SE). Unpaired t tests were used to assess the statistical significance of differences between groups. To estimate the size of the effect, mean differences and 95% confidence intervals (CI) were calculated. Correlations between variables were explored using the Spearman coefficient (rho). A p value of less than 0.05 was considered significant.
RESULTS
Clinical data
Table 1 shows the main clinical and functional characteristics of the patients included in the study. All the patients were male. By design, BMI was significantly lower in those with low body weight. Age, smoking history, and percentage of current smokers (2/7) was similar in both groups. Although both groups showed severe airflow obstruction, this was significantly greater in patients with low BMI. Gas exchange impairment was similar in both groups.
iNOS expression
As shown in fig 1, levels of iNOS expression were higher in COPD patients with low BMI (1.51 (0.29) IOD) than in those with normal body weight (0.78 (0.11) IOD, p<0.05; mean difference 0.74, 95% CI 0.04 to1.42). In our laboratory we have investigated iNOS expression in the skeletal muscle of six healthy subjects of similar age and, as expected from the literature,23 it was found to be almost absent (0.23 (0.02) IOD; data not shown).
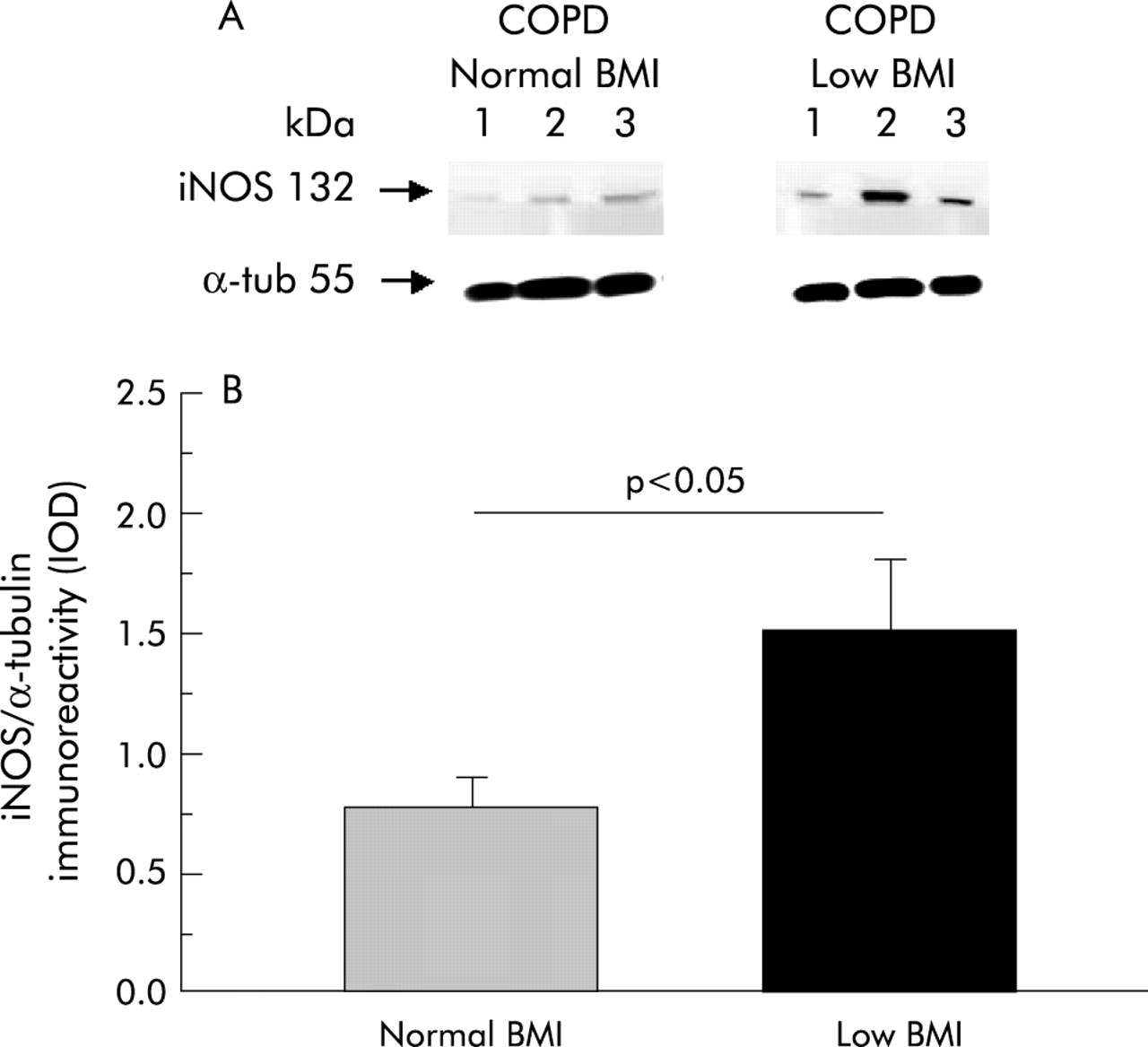
(A) Representative Western blot of iNOS in the quadriceps femoris muscle of three COPD patients with normal BMI and three patients with low BMI. (B) Mean (SE) iNOS expression (normalised by α-tubulin content) in each group.
NF-κB activation
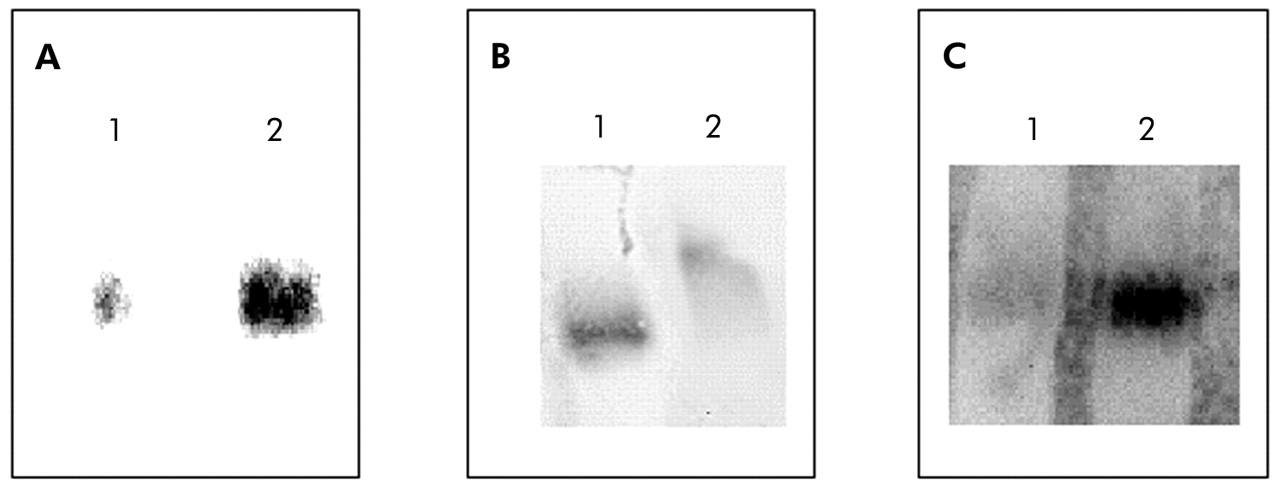
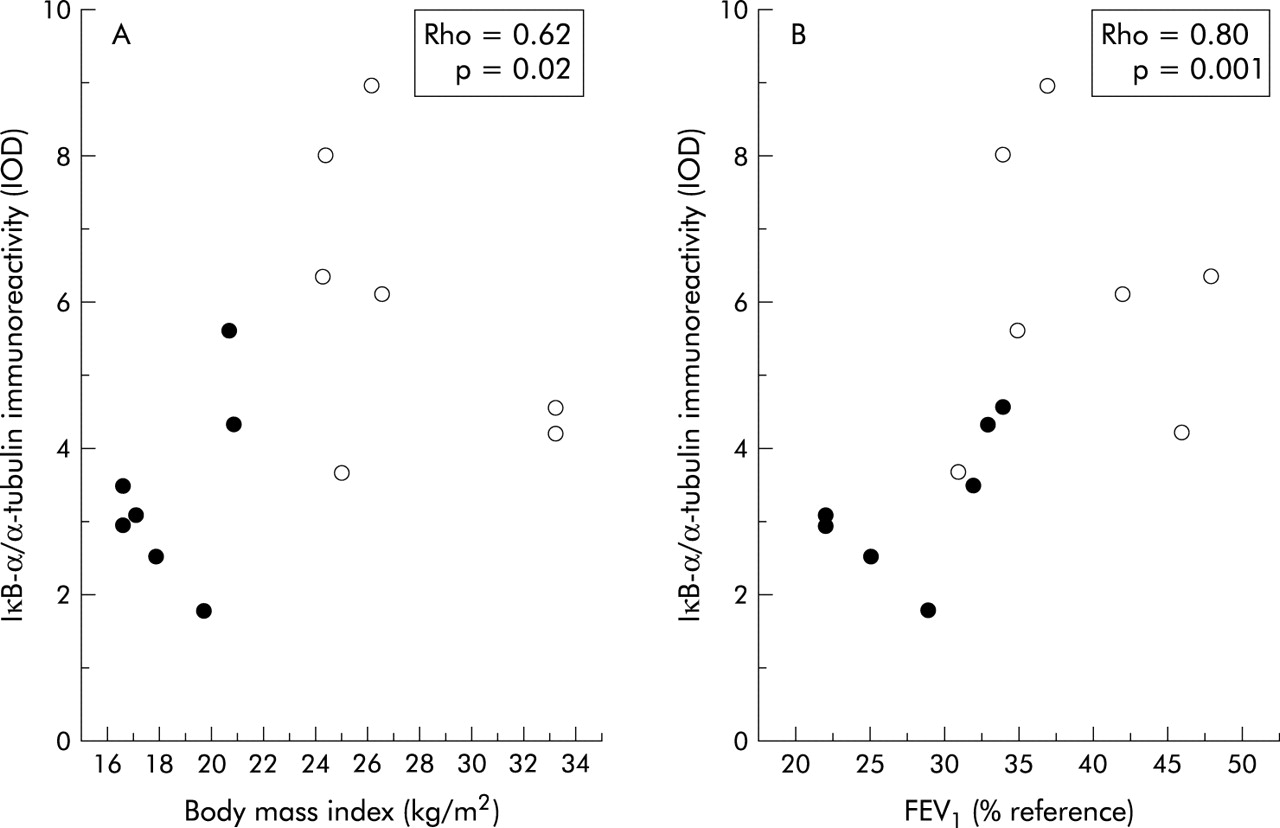
Compared with patients with normal BMI, those with low body weight showed a 30% increase in NF-κB nuclear binding (fig 2). In keeping with this observation, IκB-α immunoreactivity was significantly decreased in those with low BMI (3.37 (0.47) IOD v 5.96 (0.75) IOD, p<0.05; mean difference 2.59, 95% CI −4.53 to −0.65; fig 3). Interestingly, IκB-α immunoreactivity was significantly related to both BMI and the degree of airflow obstruction present (fig 4A and B).
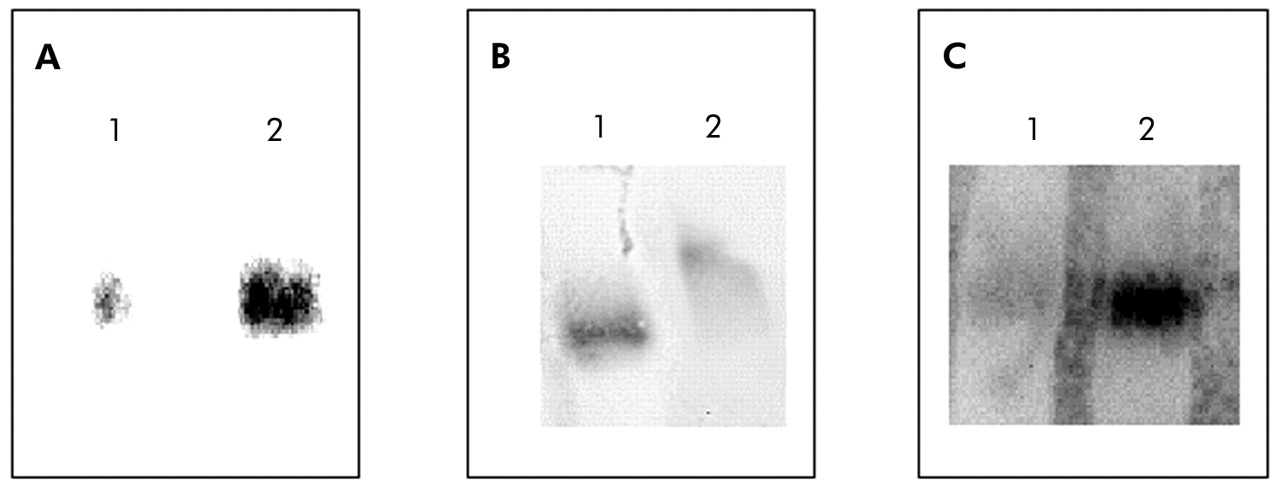
Representative electrophoretic mobility shift assays (EMSA) performed with nuclear extracts from biopsy specimens of the quadriceps femoris muscle. (A) Specificity of NF-κB binding assessed by a competition experiment with 200-fold non-labelled NF-κB oligonucleotide (lane 1) and biotin labelled NF-κB oligonucleotide. (B) Specificity of NF-κB binding assessed by a super-shift assay with nuclear extract incubated in the absence (lane 1) or presence (lane 2) of antibody against the p65 subunit of NF-κB. (C) EMSA assay performed with pooled nuclear extracts obtained from patients with normal BMI (lane 1) or low BMI (lane 2).

(A) Representative Western blot of IκB-α in the quadriceps femoris muscle of COPD patients with normal or low BMI. (B) Mean (SE) IκB-α expression (normalised by α-tubulin content) in the quadriceps femoris muscle of COPD patients with normal BMI (n = 7) or low BMI (n = 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationship between the immunoreactivity of IκB-α and (A) BMI or (B) forced expiratory volume in 1 second (FEV1) in patients with COPD. Solid circles represent patients with low BMI and open circles represent patients with normal BMI.
DISCUSSION
Weight loss is a frequent and clinically relevant problem in patients with COPD which is mainly caused by skeletal muscle atrophy,4,7 but its molecular basis is unclear.5,6 Our results contribute to a better understanding of this phenomenon by showing that activation of NF-κB and upregulation of iNOS expression occur in the skeletal muscle of patients with COPD and low BMI.
Several recent studies using a variety of techniques (including immunohistochemistry, Western blot analysis, and RT-PCR) have clearly shown that NF-κB activation and iNOS induction occur in the skeletal muscle of patients with inflammatory myopathies,24 sepsis,25 or chronic heart failure.26 To the best of our knowledge, our study is the first to show that they also occur in the skeletal muscle of patients with COPD, particularly in those with low body weight (figs 1–3). A recent study by Barreiro et al could not detect iNOS expression in the vastus lateralis muscle of 12 patients with COPD.23 However, this study assessed patients with moderate COPD (FEV1 54 (14)% predicted) and did not include patients with low BMI (25 (3) kg/m2). This clearly contrasts with our study in which patients with severe COPD (FEV1 28 (2)% predicted) and much lower BMI values (18.5 (1) kg/m2) were included. It is possible, therefore, that NF-κB activation and iNOS induction might not occur in patients with moderate disease without skeletal muscle atrophy. The correlation observed between the immunoreactivity of IκB-α and the degree of airflow obstruction (fig 4B) supports this possibility.
In humans, iNOS expression is under the control of the nuclear transcription factor NF-κB.27,28 In unstimulated cells NF-κB is localised in the cytoplasm forming a complex with its inhibitory protein IκB-α.27,28 Upon stimulation, IκB-α is cleaved from this complex and NF-κB is translocated to the nucleus (NF-κB activation) where it enhances gene transcription.27,28 We did not determine the specific factor(s) responsible for NF-κB activation and iNOS upregulation in the skeletal muscle of the patients included in our study. However, many previous studies have shown that systemic inflammation, oxidative stress, and tissue hypoxia are potent inducers of NF-κB activation and iNOS expression,11,12 and that all of them are more prevalent in patients with COPD with low body weight.5,8 It is therefore likely that they might have contributed to NF-κB activation and iNOS upregulation in our patients.
Potential implications
Nitric oxide (NO) is a free radical molecule synthesised from the amino acid l-arginine by the NO synthases (NOS).11 To date, three main NOS isoforms have been described, all of which are expressed by human skeletal muscle.23,29,30 The so-called neuronal (nNOS) and endothelial (eNOS) isoforms appear to be expressed constitutively,31,32 while the inducible isoform (iNOS) is expressed in response to a number of stimuli including cytokines, oxidants and/or hypoxia.33,34 At low concentrations, NO is involved in the regulation of numerous physiological processes in skeletal muscle32 but at high concentrations it can be cytotoxic.14 The augmented intracellular concentration of NO that will conceivably result from the induction of iNOS observed in our study can contribute to skeletal muscle atrophy—and thus to weight loss—through at least two different but not mutually exclusive pathways. On the one hand, it can enhance skeletal muscle apoptosis,14 and recent results from our laboratory support this possibility,15 while, on the other hand, it can cause protein nitrotyrosination and facilitate their preferential degradation through the ubiquitin-proteasome pathway.13 According to Barreiro et al,23 increased protein nitrotyrosination does indeed occur in the skeletal muscle of patients with COPD, and preliminary results from our laboratory (data not shown) also support this contention.
Methodological limitations
Three methodological limitations of our study deserve comment. Firstly, because of the invasive nature of the biopsy procedure used to obtain skeletal muscle samples, we studied a relatively small number of patients. This limits the direct extrapolation of our findings to the whole population of patients with COPD. Secondly, due to the relatively low protein yield of these biopsies, we determined NF-κB activation in pooled nuclear extracts. Although this is not ideal, this approach has been used by other investigators in the past.21 We are confident with our results because, on the one hand, the absolute value of the NF-κB nuclear binding increase observed (30%) is in the range of previous investigations of NF-κB activation in other respiratory diseases (44% increase in bronchial asthma21) and, on the other, IκB expression (an indirect marker of NF-κB activation28) was assessed individually. Furthermore, the observation that NF-κB activation can play a pathogenic role in the development of skeletal muscle atrophy in COPD is in keeping with recent results showing that it is also relevant in the pathogenesis of muscle decay and cachexia in patients with cancer.27 Thirdly, we did not measure NO production, so significant differences in iNOS protein expression between the two groups may not translate directly into functionally significantly higher NO levels.
Our study has shown that activation of NF-κB and upregulation of iNOS occur in the skeletal muscle of patients with COPD and low body weight. This may constitute a molecular pathway contributing to skeletal muscle atrophy in these patients.
Acknowledgments
The authors thank the patients for their willingness to participate in this project and Marga Bosch, Francisca Bauza and Angels Noguera (RNs) for their help with lung function studies.
REFERENCES
Footnotes
-
Supported in part by FIS (00/0437 and Red Respira (RTIC C3/011)), CICYT PB98-1084, SEPAR, Govern Balear and ABEMAR.
Linked Articles
- airwaves