Article Text

Abstract
Background: COPD is an inflammatory disorder characterised by chronic airflow limitation, but the extent to which airway inflammation is related to functional abnormalities is still uncertain. The interaction between inflammatory cells and airway smooth muscle may have a crucial role.
Methods: To investigate the microlocalisation of inflammatory cells within the airway smooth muscle in COPD, surgical specimens obtained from 26 subjects undergoing thoracotomy (eight smokers with COPD, 10 smokers with normal lung function, and eight non-smoking controls) were examined. Immunohistochemical analysis was used to quantify the number of neutrophils, macrophages, mast cells, CD4+ and CD8+ cells localised within the smooth muscle of peripheral airways.
Results: Smokers with COPD had an increased number of neutrophils and CD8+ cells in the airway smooth muscle compared with non-smokers. Smokers with normal lung function also had a neutrophilic infiltration in the airway smooth muscle, but to a lesser extent. When all the subjects were analysed as one group, neutrophilic infiltration was inversely related to forced expiratory volume in 1 second (% predicted).
Conclusions: Microlocalisation of neutrophils and CD8+ cells in the airway smooth muscle in smokers with COPD suggests a possible role for these cells in the pathogenesis of smoking induced airflow limitation.
- airflow limitation
- smoking
- inflammation
- chronic obstructive pulmonary disease
Statistics from Altmetric.com
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disorder of the lung defined by the presence of poorly reversible airflow limitation. The inflammatory response in COPD is characterised by a specific increase of CD8+ cells in the airway wall and of neutrophils in the airway lumen.1–4 While the inflammatory cell profile in this disease is well known,2–6 the extent to which airway inflammation is related to abnormalities in pulmonary function is still uncertain.
Of interest to the debate on the relationship between airway inflammation and pulmonary function is the specific interaction between inflammatory cells and airway smooth muscle, which may be crucial in the development of airflow limitation. A recent report showed an increased infiltration of mast cells within the bundles of airway smooth muscle in asthma, suggesting that this microlocalisation of mast cells is a key factor in the development of disordered lung function in this disease.7 However, it is unknown whether this is a specific feature of asthma or a general characteristic of obstructive airway diseases.8 To the best of our knowledge, no previous study has specifically addressed this issue in COPD. We have therefore investigated the microlocalisation of inflammatory cells within the airway smooth muscle in a group of smokers with COPD and compared the results with those in smokers with normal lung function and non-smoking controls. We focused our analysis on the peripheral airways which are considered the site responsible for the development of chronic airflow limitation in smokers.9
METHODS
Subject characterisation
Three groups of subjects undergoing lung resection for a solitary peripheral carcinoma were recruited to the study: eight smokers with symptoms of chronic bronchitis and chronic airflow limitation (smokers with COPD), 10 asymptomatic smokers with normal lung function, and eight asymptomatic non-smoking controls. According to the GOLD guidelines,1 chronic airflow limitation was defined as a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) <70% associated with an FEV1 <80% predicted, with a FEV1 reversibility of <15% after inhalation of 200 μg salbutamol. Chronic bronchitis was defined as cough and sputum production occurring on most days of the month for at least 3 months a year during the 2 years before the study. No subject had an exacerbation of his disease within the month preceding surgery. All the subjects had been free of acute upper respiratory tract infections. None of the subjects had received anti-inflammatory treatment (oral or inhaled corticosteroids) or antibiotics within the month preceding surgery or bronchodilators within the previous 48 hours. They were non-atopic (that is, negative skin tests for common allergen extracts) and had no past history of asthma or allergic rhinitis. The study conformed to the Declaration of Helsinki and informed written consent was obtained for each subject undergoing surgery. Each patient underwent an interview, chest radiography, electrocardiography, routine blood tests, skin tests with common allergen extracts, and pulmonary function in the week before surgery.
Pulmonary function tests (FEV1, FVC) and blood gas analysis were performed as previously described.10 To assess the reversibility of the airflow limitation in subjects with a baseline FEV1 <80% predicted, the FEV1 measurement was repeated 15 minutes after inhalation of 200 μg salbutamol.
Immunohistochemistry
Four to six randomly selected tissue blocks (template size 1×2×2 cm) were taken from the subpleural parenchyma of the lobe obtained at surgery, avoiding areas involved by tumour. Samples were fixed without inflation in 4% formaldehyde in phosphate buffered saline (PBS) at pH 7.2 and, after dehydration, embedded in paraffin wax. Tissue specimens were oriented and 5 μm thick sections were cut and processed for immunohistochemical analysis of inflammatory cells as previously described.11 Briefly, mouse monoclonal antibodies were used for identification of neutrophils (anti-elastase M752, Dako Ltd, High Wycombe UK), mast cells (anti-tryptase M 7052, Dako), macrophages (anti-CD68 M814, Dako), CD4+ cells (anti-CD4 M 834, Dako), and CD8+ cells (anti-CD8 M 7103, Dako). For antigen retrieval, sections to be stained for macrophages and mast cells were pretreated with 0.1% trypsin in 0.1% calcium chloride (pH 7.8) at 37°C for 20 minutes. Sections to be stained for CD8 were immersed in citrate buffer (5 mM, pH 6) and incubated in microwave oven at high power for 1 hour. Monoclonal antibody binding was detected with the alkaline phosphatase/anti-alkaline phosphatase method (APAAP kit system K670, Dako) and Fast Red substrate. A mouse monoclonal antibody was used for identification of smooth muscle actin (anti-smooth muscle actin M 0851, Dako) and the antibody binding was detected with horseradish peroxidase using diaminobenzidine substrate.
For each patient 4–9 peripheral airways, transversally cut, with an internal perimeter of less than 6 mm (corresponding to a diameter of about 2 mm) were identified. The number of mast cells, neutrophils, macrophages, CD4+, and CD8+ cells was quantified in the smooth muscle of peripheral airways. The results were expressed as the number of cells per square mm of smooth muscle area examined, as well as percentage of bronchioles infiltrated by inflammatory cells over the total number of bronchioles. A bronchiole was considered to be infiltrated by a specific cell type when there was at least one cell within the smooth muscle bundles or adjacent to them (directly contacting a smooth muscle cell). Slides were examined at magnification 630× with a light microscope (Leica DMLB; Leica, Cambridge, UK). Airway smooth muscle was identified by morphological examination and its area was calculated using a computer aided image analysis system (Casti Imaging, SC Processing, Venice, Italy). We validated our detection of airway smooth muscle by correlating the area of smooth muscle assessed by morphological examination with the area of smooth muscle assessed by a specific staining for smooth muscle actin in 10 pairs of contiguous sections; a correlation coefficient of 0.95 was obtained, indicating that smooth muscle could be correctly identified by its morphological appearance. A representative peripheral airway stained for smooth muscle actin is shown in fig 1. To avoid observer bias, the cases were coded and the measurements made without knowledge of clinical data.

Microphotograph showing a peripheral airway stained with a monoclonal antibody against smooth muscle actin. Arrows indicate smooth muscle bundles within the peripheral airway wall. Original magnification 200×.
Statistical analysis
Group data were expressed as mean (SE) or median (range) when appropriate. Differences between groups were analysed using the following tests for multiple comparisons: analysis of variance (ANOVA) and unpaired Student’s t test for clinical data and the Kruskall-Wallis and Mann-Whitney U tests for morphological data. Correlation coefficients were calculated using Spearman’s rank method. Probability values of <0.05 were accepted as significant.
RESULTS
Clinical findings
Table 1 shows the characteristics of the subjects examined. The three groups of subjects were of a similar age. Smokers with COPD had lower arterial oxygen tension (Pao2) and higher arterial carbon dioxide tension (Paco2), but the difference between the three groups of subjects did not reach statistical significance. There was no significant difference in the smoking history between smokers with COPD and smokers with normal lung function. As expected from the selection criteria, the values of FEV1 (% predicted) and FEV1/FVC (%) were different in the three groups of subjects (p<0.0001): smokers with COPD had a significantly lower FEV1 (% predicted) and FEV1/FVC ratio (%) than smokers with normal lung function and non-smokers (p<0.05). In smokers with COPD (FEV1 range 54–79% predicted, FEV1/FVC range 54–69%) the average response to bronchodilator was 5%.
Mean (SE) characteristics of study subjects
Histological findings
The total number of peripheral airways examined was 141. The mean number of airways examined per patient was 5 (0.4) in smokers with COPD, 6 (0.5) in smokers with normal lung function, and 6 (0.5) in non-smokers, with no statistical difference between groups. The airway perimeter was similar in the three groups of patients (2.278 (0.241) mm in smokers with COPD; 2.454 (0.281) mm in smokers with normal lung function, and 1.907(0.177) mm in non-smokers), indicating that we were comparing airways of similar size. The area of smooth muscle assessed per patient was similar in the three groups of patients (0.15 (0.03) mm2 in smokers with COPD; 0.13 (0.03) mm2 in smokers with normal lung function, and 0.12 (0.03) mm2 in non-smokers).
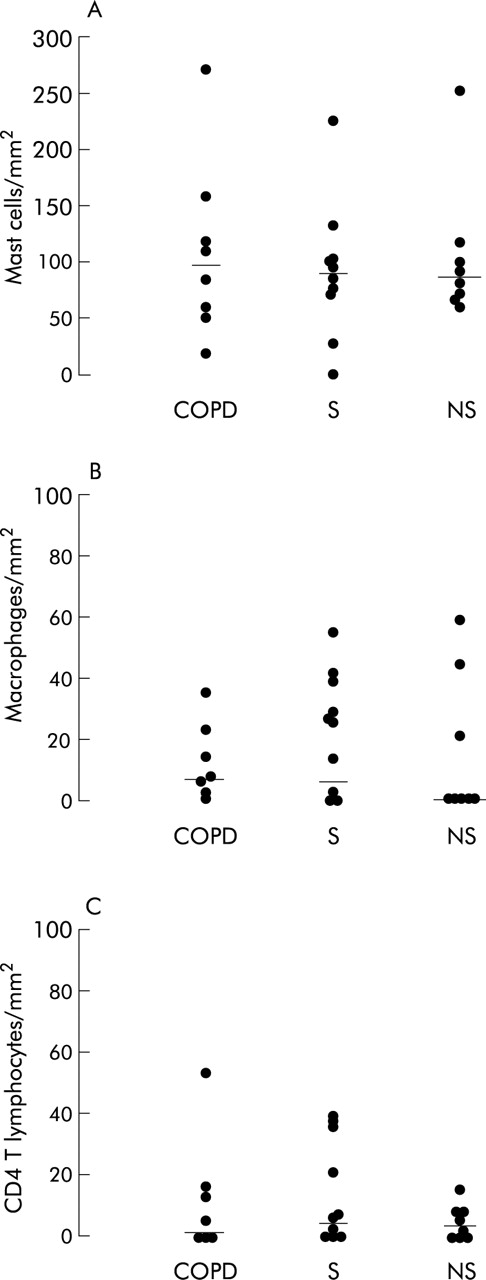
The results of cell counts in the smooth muscle of peripheral airways are shown in figs 2–4. The number of neutrophils and CD8+ cells infiltrating the airway smooth muscle was different in the three groups of patients (p = 0.005 and p = 0.05, respectively). Smokers with COPD had an increased number of neutrophils within the airway smooth muscle compared with non-smokers (median 53 (range 19–93) v 9 (0–38) cells/mm2; p = 0.002). Smokers with normal lung function also had a higher number of neutrophils than non-smokers, although the increase was less (median 23 (range 0–114) cells/mm2; p = 0.046). The number of CD8+ cells was higher in smokers with COPD than in non-smokers (median 19 (range 0–33) v 0 (0–15); p = 0.036), while the number of mast cells, macrophages and CD4+ cells did not differ between the three groups of subjects examined (table 2). The mean “between airways” coefficient of variation was 1.29 for neutrophils, 1.05 for CD8+ cells, 1.18 for CD4+ cells, 1.03 for macrophages, and 1.03 for mast cells.
Median (range) cell counts per mm2 in airway smooth muscle

Individual counts for (A) neutrophils and (B) CD8+ cells within the smooth muscle of peripheral airways in smokers with COPD (COPD), smokers with normal lung function (S), and non-smokers (NS). The results are expressed as number of cells per square mm of smooth muscle examined. Horizontal bars represent median values.

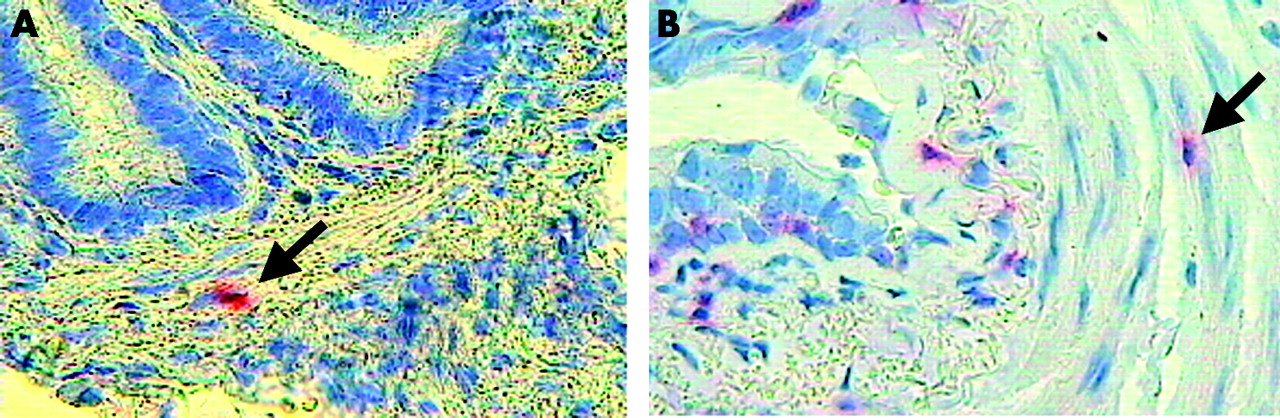
Microphotograph showing (A) neutrophils (arrow) and (B) CD8+ cells (arrow) within the airway smooth muscle of peripheral airways of smokers with COPD. Immunostaining with monoclonal antibodies anti-human neutrophil elastase and anti-CD-8. Original magnification 630×.

Individual counts for (A) mast cells, (B) macrophages, and (C) CD4+ cells within the smooth muscle of peripheral airways in smokers with COPD (COPD), smokers with normal lung function (S), and non-smokers (NS). Results are expressed as number of cells per square mm smooth muscle examined. Horizontal bars represent median values.
When the results were expressed as a percentage of bronchioles infiltrated by inflammatory cells over the total number of bronchioles, the percentage of bronchioles infiltrated by neutrophils was different in the three groups of patients (p = 0.04). Smokers with COPD (45% (range 17–86%)) and smokers with normal lung function (53% (0–100%)) had an increased percentage of bronchioles infiltrated by neutrophils compared with non-smokers (20% (0–44%); p = 0.021 and p = 0.041, respectively). Moreover, smokers with COPD showed a trend toward an increased percentage of bronchioles infiltrated by CD8+ cells compared with non-smokers (19% (0–33%) v 0% (0–15%); p = 0.058). The percentage of bronchioles infiltrated by mast cells, macrophages, and CD4+ cells was similar in the three groups.
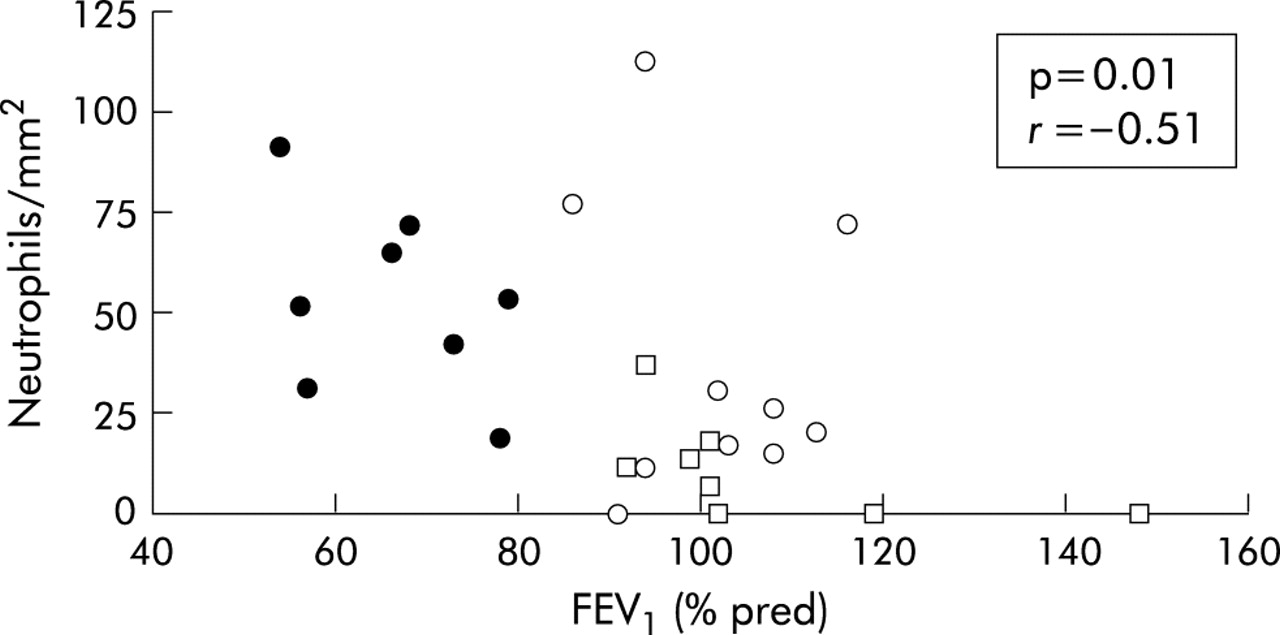
When all the subjects were considered together, the number of neutrophils in the airway smooth muscle showed a negative correlation with both FEV1 % predicted (r = −0.51; p = 0.01, fig 5) and FEV1/FVC % (r = −0.42; p = 0.036). No significant correlations were observed between other inflammatory cells and the degree of airflow limitation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationship between number of neutrophils within the smooth muscle of peripheral airways and forced expiratory volume in 1 second (FEV1) % predicted in all subjects in the study (r = −0.51, p = 0.01). Closed circles indicate smokers with COPD, open circles indicate smokers with normal lung function, open squares indicate non-smokers.
DISCUSSION
This study shows that, in peripheral airways of smokers with COPD, there is an increased number of neutrophils and CD8+ cells infiltrating the smooth muscle. Even smokers with normal lung function had a neutrophilic infiltration within the airway smooth muscle, although to a lesser extent.
Both CD8+ cells and neutrophils are known to be important in the pathogenesis of chronic airflow limitation.1–4 Indeed, a CD8 immune response involving the whole tracheobronchial tree has been described in smokers with COPD, so the finding of an increased number of CD8+ cells in the peripheral airway smooth muscle was not unexpected. Conversely, although a prominent neutrophilia has been described in bronchoalveolar lavage fluid and induced sputum from smokers with COPD,4,12,13 most of the studies examining the inflammatory cells infiltrating the peripheral airway wall (not focused on smooth muscle) failed to find an increased number of neutrophils.3,5,6 We were therefore surprised by the finding of a neutrophilia in the smooth muscle compartment which may have important functional consequences.
In obstructive airway diseases the extent to which airway inflammation is related to pulmonary function is still a controversial issue and, in this context, the specific interaction between inflammatory cells and airway smooth muscle may be of interest. A recent study reported an increased number of mast cells in the airway smooth muscle of patients with asthma compared with patients with eosinophilic bronchitis,7 a condition characterised by normal lung function despite the presence of an airway eosinophilia similar to that present in asthma. The only difference seen in the pathology between the two conditions was the number of mast cells in the airway smooth muscle, which suggests that microlocalisation of mast cells within the bundles of smooth muscle represents a key factor in the development of the disordered lung function in asthma. However, given the absence of a control group of subjects with other obstructive lung diseases, this study does not show whether such infiltration is a specific feature of asthma or a general characteristic of obstructive airway diseases.8 Following this reasoning, our study contributed to the debate by showing that, unlike in asthma, neutrophils and CD8+ cells (but not mast cells) are increased in the airway smooth muscle of smokers with COPD.
In the present study neutrophils were increased not only in smokers with airflow limitation but, to a lesser extent, in smokers with normal lung function, which suggests that smoking itself may play a role in this inflammatory response. In this context, a recent study by Berger and coworkers reported that the neutrophil infiltration of airway smooth muscle in smokers was related to the amount of air trapping measured by CT scanning.14 It is conceivable that cigarette smoking promotes an early recruitment of neutrophils within the smooth muscle which progresses with the development of airflow limitation. The negative correlation observed in our study between neutrophilic infiltration and FEV1 might support this hypothesis. We should acknowledge, however, that this correlation pooled together data from three combined groups of patients, thus introducing a potential bias which may limit its value.
Unlike neutrophils, CD8+ cells were increased only in smokers with COPD, indicating that these cells are selectively recruited when the disease is established. This finding is in agreement with previous reports showing CD8+ cell infiltration in several lung compartments in smokers with COPD but not in smokers with normal lung function.3,11 Although it is usually thought that CD8+ cells are likely to represent cytotoxic T lymphocytes, other cells (particularly NK cells) may also express this marker.
The establishment of airflow limitation in smokers is paralleled by the development of structural abnormalities in peripheral airways, including an increased area occupied by smooth muscle.3,15 The major functional consequence of the increase in smooth muscle mass is that, in airways with thickened walls, the same degree of smooth muscle shortening may cause considerably greater luminal narrowing than in normal airways.16 It is conceivable that chronic airway inflammation could contribute to changes in the structure and contractility of airway smooth muscle, possibly through the action of inflammatory mediators and cytokines. Indeed, it has been shown that altered peripheral airway smooth muscle function is present in smokers with COPD.17 Interestingly, interleukin 8, a potent neutrophil chemoattractant which has been shown to be increased in COPD,4 is able to induce smooth muscle proliferation and constriction.18,19 Further studies investigating the effects of neutrophil and CD8 derived cytokines are needed to clarify the mechanisms of interaction between inflammatory cells and airway smooth muscle cells in COPD.
Although a high number of neutrophils is consistently found in the airway lumen of smokers with COPD,4,12,13 these cells are scanty in the airway submucosa of these subjects.2,3,5,6,20 A possible explanation for this discrepancy is that different microenvironments within the airways may influence the inflammatory response. By showing a microlocalisation of neutrophils within the smooth muscle bundles, this study suggests that these cells are recruited at sites where inflammatory mediators can directly act on target effectors such as airway smooth muscle. This hypothesis is supported by our previous observation that smokers with COPD have a neutrophilic infiltration in the bronchial glands,21 where these cells can act as potent secretagogues.22
Increased infiltration of neutrophils and CD8+ cells was present in smokers with COPD when the results were expressed both as number of cells per mm2 smooth muscle and as a percentage of bronchioles infiltrated by inflammatory cells over the total number of bronchioles examined. This indicates that the extent of smooth muscle inflammation, as well as the intensity, was increased in these subjects.
The absolute number of mast cells, even if not different between groups, was largely higher than the number of other inflammatory cells infiltrating the airway smooth muscle. These results agree with those of Carroll and coworkers23 who found that mast cells were the predominant cells within the airway smooth muscle layer in both asthmatic and non-asthmatic subjects, with a mast cell density similar to that observed in our study. Furthermore, in our study the number of mast cells was greater than that recently reported by Brightling and coworkers.7 However, there are significant differences between the two studies which may have influenced the results. For example, we examined peripheral airways while Brightling et al examined central airways, and there is evidence that mast cell density is higher in peripheral than in central airways.23,24 Moreover, Brightling and coworkers used glycol methacrylate embedded sections from asthmatic patients whereas we used paraffin embedded sections from patients with COPD. Conclusions cannot therefore be drawn on the direct comparison in mast cell density between the two studies.
A confusing element in any study performed on surgically resected specimens from patients with lung cancer is that the presence of cancer itself may influence the results. However, surgical specimens are the only specimens that allow for the examination of peripheral airways in patients well characterised in terms of pulmonary function. These samples are extremely important as peripheral airways are the major site responsible for the development of chronic airflow limitation in smokers.9 Moreover, because we examined only tissue away from the tumour site and included subjects with lung cancer in the control groups, we are confident that our finding of a microlocalisation of neutrophils and CD8+ cells within airway smooth muscle in COPD is valid.
In conclusion, in peripheral airways of smokers with COPD there is an increased number of neutrophils and CD8+ cells within the bundles of smooth muscle. Smokers with normal lung function also have a neutrophilic infiltration within the smooth muscle, but to a lesser extent. These findings suggest a possible role for the interaction between inflammatory cells and airway smooth muscle in the pathogenesis of smoking induced airflow limitation.
REFERENCES
Footnotes
-
Supported by Italian Ministry of University and Research and University of Padova (grant CPDG 035929).
Linked Articles
- airwaves