Article Text

Abstract
The pathogenesis of airway obstruction in patients with obstructive sleep apnoea/hypopnoea syndrome is reviewed. The primary defect is probably an anatomically small or collapsible pharyngeal airway, in combination with a sleep induced fall in upper airway muscle activity.
- obstructive sleep apnoea
Statistics from Altmetric.com
Obstructive sleep apnoea/hypopnoea syndrome (OSAHS) is a common disorder which affects 2–4% of middle aged women and men in the United States.1 This disorder is characterised by recurrent sleep induced collapse of the pharyngeal airway2 leading to hypoxaemia and hypercapnia, with arousal from sleep being required to re-establish airway patency. While the pathophysiology of this disorder remains incompletely understood, much progress has been made since the last review of this topic was published in this journal.3 We believe that most evidence supports the concept that patients with OSAHS have an anatomical predisposition to airway collapse. However, during wakefulness, protective mechanisms maintain the patency of the pharyngeal airway by increasing the activity of pharyngeal dilator muscles. These protective mechanisms fail during sleep, with subsequent collapse of the pharyngeal airway behind the palate, tongue, or both. In this review we consider the current state of knowledge regarding the pathogenesis of airway obstruction in patients with OSAHS, emphasising those theories that have the most experimental support.
MECHANICS OF THE PHARYNX
The pharyngeal airway is a complex structure (fig 1) whose functions include respiration, speech, and swallowing. Thus, the evolution of the structure and function of the pharynx has required “trade offs” between these competing functions. It is hypothesised that the evolution of speech in man, which requires substantial mobility of the larynx, led to the loss of rigid support of the hyoid bone which is present in most mammals. The human pharyngeal airway is therefore largely dependent on muscle activity to maintain its patency. However, the dependence of individual subjects on this pharyngeal dilator muscle activity is probably a product of the intrinsic anatomy and collapsibility of their airway. Individuals with an anatomically large airway may be less dependent on muscles to maintain airway patency than those with a small airway. When pharyngeal collapse does occur, it generally does so in the velopharynx (behind the soft palate), the oropharynx (from the tip of the soft palate to the epiglottis), or both.4,5

Anatomical representation of the upper airway and the important muscles controlling airway patency. In patients with apnoea, airway collapse typically occurs behind the palate (velopharynx), the tongue (oropharynx), or both.
The human pharynx can be modelled as a collapsible tube, the patency of which can be described using a “balance of pressures” concept.2,6 The size of the upper airway depends on the balance between those forces that would collapse the airway (such as negative intraluminal pressure and increased tissue (extraluminal) pressure) and those that maintain airway patency (contraction of pharyngeal dilator muscles, see below). The transmural pressure of the pharynx (Ptm) is thus equal to the pressure in the lumen (Pl) minus the surrounding pressure in the tissue (Pti), with the airway lumen becoming smaller as Ptm decreases. The change in area for a given change in pressure describes the effective elastance of the pharynx. The Ptm at which the area of the pharynx equals zero is the closing pressure of the pharynx. A number of studies have used this model of pharyngeal mechanics to look at differences in the mechanical properties of the pharynx in normal controls and subjects with OSAHS, and generally report a higher (less negative or even positive) closing pressure in those with OSAHS. Schwartz and colleagues measured the critical closing pressure (Pcrit, pressure at zero flow) during sleep in patients with OSAHS and controls.7 These studies8 have shown that normal subjects typically have Pcrit values below −8 cm H2O while those with mild OSAHS/snoring have a slightly negative Pcrit value and those with severe disease have a Pcrit of >0. This again suggests an anatomically smaller airway in patients with apnoea that is more collapsible during sleep when muscle activity may be low.
ANATOMY OF THE PHARYNX
There is abundant evidence to suggest anatomical differences in the pharyngeal airway between patients with OSAHS and controls. Cephalometric x rays were among the earliest techniques used to image the pharyngeal airway and showed that patients with OSAHS have a reduction in the length of the mandible, an inferiorly positioned hyoid bone, and a retroposition of the maxilla.9 Using more sophisticated imaging techniques including CT scanning, acoustic reflection and, most recently, MRI, a number of investigators have shown that patients with apnoea have a smaller airway lumen than controls.5,10–13 Studies by Schwab et al have also indicated a number of soft tissue abnormalities in patients with apnoea including an increase in the volume of the tongue, soft palate, parapharyngeal fat pads, and the lateral walls surrounding the pharynx.13 In addition to a difference in size, a number of studies have shown a difference in airway shape, with apnoeic subjects having an airway with its long axis directed anterior-posterior rather than laterally. Some have suggested that this airway orientation in patients with apnoea may place pharyngeal dilator muscles at a relative mechanical disadvantage, decreasing their ability to maintain pharyngeal patency.14 Recent data also suggest that airway length may be an important anatomical variable in determining patency. Sforza et al found that the distance between the hyoid bone and the mandibular plane predicted collapsing pressure (Pcrit) in a group of men with OSAHS.15 Verin et al found this cephalometric variable to be the best predictor of upper airway resistance in awake apnoea patients and controls.16 Data from our laboratory using finite element analysis modelling suggest that increasing airway length renders the pharyngeal airway more susceptible to collapse.17 A number of anatomical variables therefore contribute to increased pharyngeal collapsibility in patients with apnoea.
The observed variability in airway size is probably determined by genetic influences on bony structure, tongue size, and tonsillar tissue as well as acquired factors such as obesity. Obesity may affect pharyngeal size by direct deposition of fat around the airway or by altering muscle orientation and function. A number of factors therefore combine to determine airway size which, importantly, impacts on the propensity for pharyngeal collapse and apnoea during sleep.
Although the studies indicating anatomical differences are compelling, most were performed during wakefulness when airway luminal size reflects both pharyngeal anatomy and dilator muscle activation. Conclusions regarding pure anatomy from these images are therefore difficult. However, a relatively recent study by Isono et al18used endoscopic techniques to assess pharyngeal airway size in both patients with apnoea and controls under general anaesthesia with full muscle paralysis and found a smaller pharyngeal airway and increased airway collapsibility in apnoea patients than in controls.18 True anatomical differences must therefore be present.
PHARYNGEAL MUSCLE FUNCTION AND THE EFFECT OF SLEEP
Although the muscles responsible for maintaining patency of the upper airway and the mechanisms controlling their function are incompletely understood, certain generalisations can be made. The muscles of primary importance fall into three groups: (1) muscles influencing hyoid bone position (geniohyoid, sternohyoid), (2) the muscle of the tongue (genioglossus), and (3) the muscles of the palate (tensor palatini, levator palatini). The activity of many of these muscles is increased during inspiration, thus stiffening and dilating the upper airway and acting to counteract the collapsing influence of negative airway pressure.19 These are referred to as inspiratory phasic upper airway muscles, with the genioglossus being the one best studied. The activity of these muscles is substantially reduced (although not eliminated) during expiration (tonic activation) when pressure inside the airway becomes positive and there is less tendency for collapse. Other muscles such as the tensor palatini do not consistently have inspiratory phasic activity but, instead, maintain a relatively constant level of activity throughout the respiratory cycle.20 These are called tonic or postural muscles and are also thought to play a role in the maintenance of airway patency. These two types of pharyngeal muscles are probably controlled by groups of neurones within the brainstem that have different firing patterns relative to the respiratory cycle.
The activity of the pharyngeal dilator muscles is carefully controlled by a number of variables (fig 2). Firstly, motor nuclei controlling these muscles receive input from central respiratory pattern generating neurones located in the ventral medulla. It is probably these neurones that lead to the previously described pattern of activation of the genioglossus (inspiratory phasic activation) as this muscle becomes active before the onset of diaphragmatic contraction or inspiratory airflow, thus “preparing” the upper airway for negative pressure. Secondly, standard respiratory stimuli (rising Pco2 and falling Po2) can augment the activity of these muscles, probably working through the respiratory premotor neurones. Thirdly, as in other components of the respiratory system, a “wakefulness” drive to these muscles may exist and may well be mediated through the state sensitive (decreased activity during sleep) neural systems discussed below.
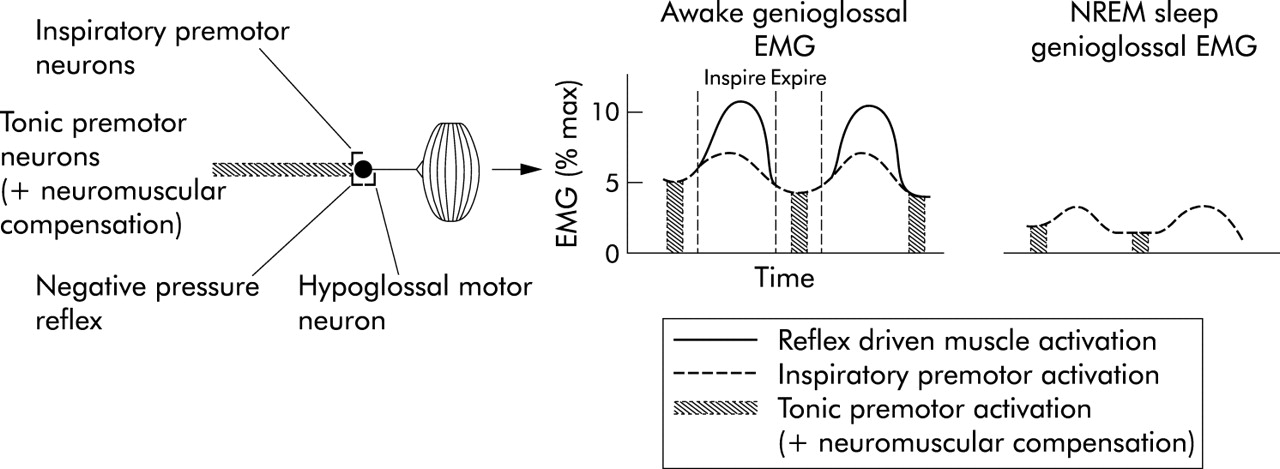
Diagram showing the three major inputs to the genioglossal muscle and muscle activation in a patient with obstructive apnoea during wakefulness and during NREM sleep. During sleep there is loss of both tonic premotor input (and neuromuscular compensation) and reflex driven muscle activation leading to a large decrement in EMG and ultimately airway collapse.
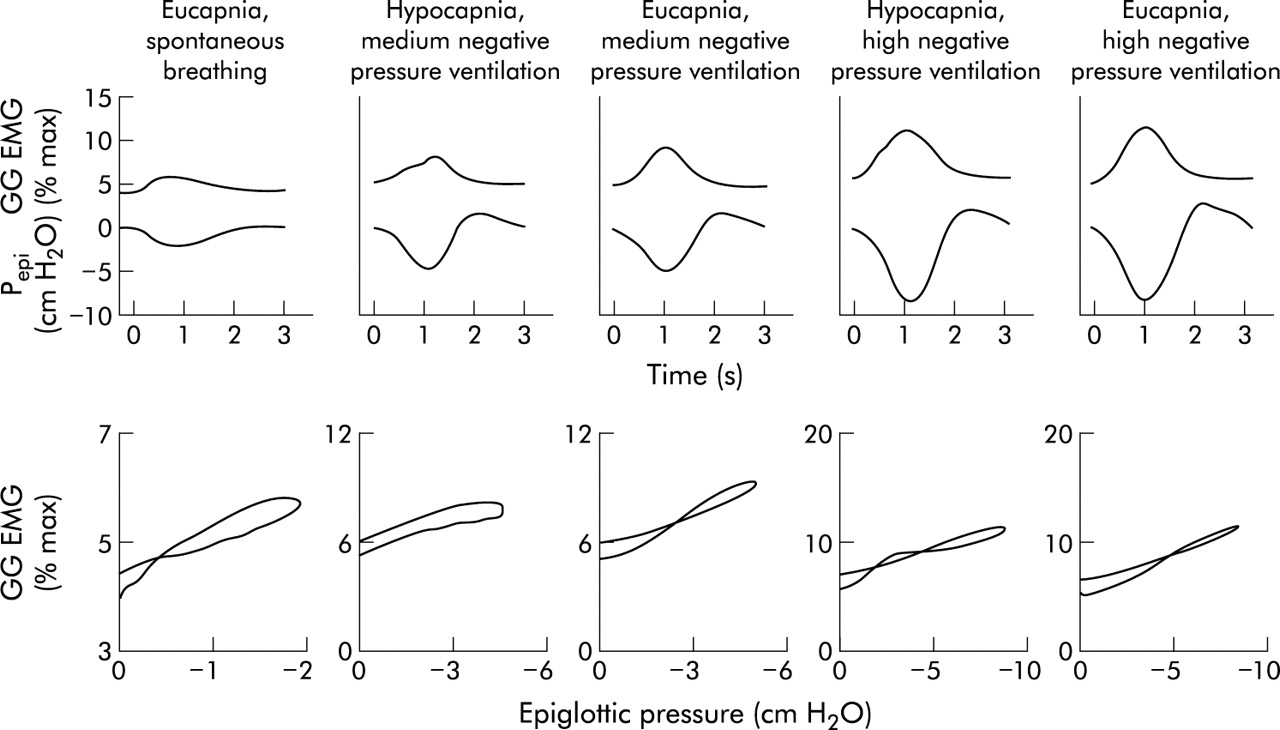
Finally, several lines of evidence suggest that negative intrapharyngeal pressure is the most important local stimulus to activation of the pharyngeal muscles during wakefulness. A pulse of negative pressure applied to the pharyngeal airway leads to robust activation of these muscles in both animals and humans.21–23 That this activation is mediated locally is shown by the fact that topical anaesthesia substantially reduces this reflex.24 In addition, if an individual is ventilated using an iron lung (negative pressure ventilation) such that respiratory effort is largely eliminated, there remains a remarkably linear relationship between the negative pressure in the airway (Pepi) and genioglossal EMG activation (GG EMG, fig 3). This relationship between Pepi and GG EMG is quite constant within a given individual across a wide range of pressures, CO2 levels, and airflows.25 Thus, even in the absence of central respiratory modulation, these muscles are able to respond on a millisecond by millisecond basis to collapsing negative pressure, thereby maintaining airway patency.

{kind=link}
{kind=link}
{kind=link}
Relationship between negative epiglottic pressure (Pepi) and genioglossal EMG (GG EMG) during inspiration across a range of conditions. A close relationship is seen between changes in Pepi and GG EMG during inspiration which is not altered by changes in CO2 levels (top panel). The slope of this relationship is fairly constant (lower panel).
As stated above, the activity of the pharyngeal dilator muscles is, during wakefulness, tightly controlled in an attempt to maintain pharyngeal patency. In patients with apnoea with an anatomically small and more collapsible airway, activation of this negative pressure reflex should lead to increased activation of the pharyngeal muscles to assure airway patency. This has now been shown for both the genioglossus and the tensor palatini muscles.26,27 Similar observations have been made in the English bulldog, a naturally occurring animal model of OSAHS.28 That negative pressure drives this augmented activity in patients with apnoea is shown by the fact that application of positive pressure (CPAP) reduces GG EMG to nearly normal levels in patients with apnoea but has little effect in controls (suggesting that this reflex is not substantially active during quiet breathing in controls).27 It has also been shown that the increased genioglossal activity in the patients with apnoea has two components. Firstly, the negative pressure in the upper airway of patients with apnoea is greater (more negative) than that of controls. This is almost certainly a product of the smaller pharyngeal lumen and the need for greater intrapharyngeal pressure to generate adequate airflow. This increased negative pressure yields increased phasic activity of the genioglossus. However, the slope of the relationship between GG EMG and Pepi does not differ between apnoea patients and controls. Secondly, the tonic (basal, non-inspiratory) activity of these muscles is also greater in patients with apnoea.26 The mechanism(s) underlying the increase in tonic activation are as yet unclear, but may represent a plasticity of the neural system involved. Thus, the airway muscles compensate very precisely for the deficient anatomy of apnoea patients while awake and ventilation is maintained.
The importance of the sleep-wake state in the pathogenesis of OSAHS is highlighted by the fact that disordered breathing events only occur during sleep, even in patients with the most severe apnoea. In control subjects sleep has a noticeable effect on pharyngeal muscle function and mechanics; the transition from wake to sleep is associated with an initial small fall in the inspiratory phasic activity of muscles such as the genioglossus which then recovers to waking (or slightly greater) levels within a few breaths.29 However, muscles with a primarily tonic activation pattern (such as the tensor palatini) lose activity at sleep onset which continues to fall as sleep deepens, reaching levels of 20–30% of waking values during stage 4 sleep.20 The differential effect of sleep on the activity of these two types of muscles (phasic versus tonic) almost certainly relates to differential effects on the brainstem neurones controlling their activity. By recording brainstem neurones across sleep-wake states in cats, Orem et al have shown that neurones with activity closely related to the respiratory cycle (phasic neurones) largely maintained their activity during sleep, while neurones whose activity pattern is not clearly related to the respiratory cycle (tonic neurones) have large decrements in activity.30 Despite the maintenance of activity in many pharyngeal dilator muscles, sleep is associated with a significant rise in upper airway resistance, even in normal subjects.31,32 This may be due to a decrement in the activity of tonic muscle as noted above.
Sleep is associated with a significant reduction in multiple neural reflex mechanisms including postural, spindle-driven reflexes. This also appears to be the case for negative pressure reflexes in the upper airway. As stated above, during wakefulness both rapid pulses of negative pressure and slow phasic increases in negative pressure lead to marked activation of the genioglossal muscle (fig 3). Many studies have suggested that, during NREM sleep, this negative pressure reflex is substantially diminished or lost completely.33 Furthermore, the relationship between changes in Pepi and GG EMG is much lower during sleep.34 During passive negative pressure ventilation in the iron lung there is a marked reduction in the GG EMG response to negative pressure during NREM sleep compared with wakefulness, leading to a rise in airflow resistance and, in some normal subjects, the development of airway obstruction.35
If the augmented pharyngeal muscle activity in patients with apnoea is driven by these neural reflex mechanisms, then sleep onset should be associated with a greater reduction in pharyngeal muscle activity in these patients. In fact, this seems to be the case. Studies of genioglossal muscle activity at sleep onset suggest that patients with apnoea have a much greater reduction in GG EMG than normal subjects.36 This is interpreted as a loss of the neural reflex mechanisms that drive neuromuscular compensation, which are activated in the apnoea patient but not in normal subjects. This decrement in muscle activity in patients with apnoea who have an anatomically small and more collapsible airway leads to pharyngeal collapse resulting in apnoea (fig 2). The state dependent neural mechanism responsible for the loss of these neural reflex mechanisms is unclear at this time. However, virtually all neural systems driving wakefulness (serotonin, noradrenaline, acetylcholine, histamine, and orexin) project monosynaptically to the hypoglossal motor nucleus (controlling the genioglossus) and all are excitatory. Thus, when these neural systems lose activity during sleep, it is not surprising that muscle activity falls.
OTHER FACTORS INVOLVED IN THE PATHOGENESIS OF APNOEA
Lung volume
An association between changes in lung volume and the size of the upper airway has been described for both animals and humans, such that upper airway size increases at higher lung volumes.37,38 This effect is probably due to increased “tracheal tug” leading to an increase in upper airway size and decreased airflow resistance.39 It has also been shown that patients with OSAHS have a greater change in upper airway dimensions with changes in lung volume—that is, a greater “lung volume dependence” of upper airway size.40 The importance of this effect is unknown, but preliminary data from our laboratory suggest that passive reductions in lung volume during NREM sleep are associated with increased airflow resistance and collapsibility of the upper airway, despite an associated increase in genioglossal muscle activation (M Stanchina, personal communication). A decrement in end expiratory lung volume associated with NREM sleep occurs in normal subjects. Whether there is an exaggerated fall in lung volume at sleep onset in patients with apnoea which might further predispose to airway collapse is unknown.
Ventilatory stability
The role of ventilatory control mechanisms in the pathogenesis of OSAHS is less clear as consistent differences in standard measures of respiratory drive have not been shown in patients with OSAHS or controls.41 However, several studies indicate that instability of respiratory control can lead to periodic breathing and that compromised airway patency (complete or partial obstruction) can occur at the nadir of the ventilatory cycle.42 As a result, instability of respiratory control may contribute, in some individuals, to the development of OSAHS. However, there has not been convincing evidence that patients with OSAHS have such instability of respiratory control. The best current measure of respiratory instability is “loop gain”, which is a measurement of the tendency of the ventilatory control system to amplify respiration in response to a stimulus or perturbation. Ventilation will become unstable if the loop gain is >1 or will stabilise in response to this same perturbation if the loop gain is <1. Younes et al have recently developed a method for measuring loop gain during sleep using proportional assist ventilation43 and have shown that some patients with sleep apnoea have an increased loop gain and thus a more unstable respiratory control system.44 Ventilatory instability is likely to be most important in apnoea patients with only a mild pharyngeal anatomical deficiency. If these individuals have a high loop gain, then the fluctuating upper airway resistance which occurs at sleep onset may be sufficiently destabilising that ventilation will begin to cycle leading to further loss of pharyngeal muscle activation and frank airway collapse.
PROGRESSION OF SLEEP APNOEA/HYPOPNOEA
While good data are available on the prevalence of OSAHS, there is less information available on the progression of the disease. However, several studies suggest that the severity of apnoea worsens over time. In the Wisconsin Sleep Study the mean apnoea-hypopnoea index (AHI) increased from 2.6 to 5.1 events per hour over 8 years. The increase in AHI was substantially greater in obese individuals and in those who reported habitual snoring at baseline. The reason for worsening severity of apnoea over time is incompletely understood, but several factors may play a part. Firstly, snoring and repeated upper airway occlusion can lead to oedema and swelling of upper airway soft tissue structures which may contribute to further narrowing of the upper airway, making it more susceptible to collapse. This oedema has been shown to be reversible with nasal CPAP therapy and may explain why the severity of apnoea is reduced on a single night without CPAP.
Others have suggested that pharyngeal muscle dysfunction may contribute to the development or progression of apnoea for several reasons. Repeated contraction of upper airway dilator muscles during experimental airway occlusion in cats leads to eccentric contraction of the muscle (contraction during active muscle lengthening), a pattern known to cause muscle injury.19 Repeated vibratory trauma which occurs during snoring is another potential source of injury or remodelling of the upper airway muscles or nerves. This concept is supported by the fact that patients with OSAHS have an impaired ability to detect sensory stimuli in the upper airway which can be at least partially reversed with nasal CPAP.45 However, whether important pharyngeal muscle injury occurs in sleep apnoea and whether such injury is pathophysiologically important is controversial. Using the English bulldog model of OSAHS, Petrof and colleagues have identified a number of changes in the upper airway musculature. These include a change in fibre type (increased fast twitch fibres) as well as signs of muscle injury and fibrosis.46,47 Others have shown similar changes in palatal muscle specimens from humans.48,49 However, studies by Series et al found no evidence of myopathy in the musculus uvulae of patients with apnoea but an increase in both anaerobic metabolism and force generating capacity.50 The role of pharyngeal muscle injury or dysfunction in the pathogenesis or progression of apnoea has therefore yet to be completely defined.
CONCLUSIONS
This review suggests that the primary defect in OSAHS is an anatomically small or collapsible pharyngeal airway. During wakefulness, neuromuscular compensatory systems function to increase the activity of the pharyngeal dilator muscles, thus preserving airway patency. However, this reflex driven augmented muscle activity is lost at sleep onset, and collapse of the pharyngeal airway occurs. The associated hypoxaemia and hypercapnia drive increasing respiratory effort, and ultimately arousal from sleep occurs, thus re-establishing airway patency and ventilation. Once the patient returns to sleep the cycle begins again. The patient thus suffers the consequences of repeated sleep disruption as well as recurrent hypoxaemia and hypercapnia.