Article Text

Statistics from Altmetric.com
New insights into the pathophysiology of acute chest syndrome (ACS) have highlighted potential therapeutic strategies including the use of nitric oxide. A multidisciplinary approach to the management of patients admitted with a sickle cell crisis is needed to prevent progression to ACS and need for mechanical ventilation.
CASE REPORT
A 21 year old Afro-Caribbean man with known sickle cell disease (SCD) was admitted to hospital with painful chest, thighs, and generalised abdominal pain. This was his second admission to hospital with a sickle cell crisis. He was on no regular medications apart from analgesia taken at home during crises. Apart from anaemia, there was nothing abnormal to find on examination. The haemoglobin (Hb) was 7.5 g/dl (normal for him), white cell count 22.5 × 109, and the electrolytes were normal apart from a slightly raised C-reactive protein (CRP) level of 30 mg/l. Radiographs of his abdomen and thighs were normal, but his chest radiograph showed a small degree of basal atelectasis bilaterally. His oxygen saturation was 96% on air. He was treated with a subcutaneous opiate infusion using a syringe pump, intravenous fluids, oxygen, and encouraged to drink.
Over the next 24 hours his pain was not well controlled and required an increasing dose of opiates. He became pyrexial (38.5°C), his oxygen saturation fell to 92% on air, and antibiotics to cover community acquired pneumonia were commenced. On the third day, however, he became more drowsy with arterial blood gases of pH 7.35, Pao2 13.5 kPa, and Paco2 7.5 kPa on 40% oxygen. A repeat chest radiograph showed new infiltrates in both lower zones and a diagnosis of acute sickle chest syndrome (ACS) was made. Despite an exchange transfusion, he continued to deteriorate and was eventually intubated and ventilated. On intensive care his Fio2 was reduced from the initial 50% to 28% within 24 hours. Sputum samples obtained by tracheobronchial suction showed no significant bacterial growth, but his CRP had risen to 150 mg/l so the antibiotic spectrum was broadened. After 3 days of mechanical ventilation his chest radiograph showed significant clearing of the lower zones, he was extubated without incident, and discharged from hospital after a further few days. Subsequent atypical respiratory serological examination did not show any rise in titres.
DISCUSSION
ACS consists of a combination of signs and symptoms including dyspnoea, chest pain, fever, cough, multifocal pulmonary infiltrates on the chest radiograph, and a raised white cell count. It is a form of lung injury that can progress to adult respiratory distress syndrome (ARDS). It is estimated that half of all patients with sickle cell anaemia will develop ACS at least once in their lives, and ACS is the second most common cause of admission after painful vaso-occlusive crises. The most recent statistics from the USA suggest that 13% of patients with ACS require mechanical ventilation with a mortality rate of 3%, mostly affecting adults1; the average length of stay was 10.5 days. ACS is the most common cause of death in these patients2–4 and may be the cause of the chronic pulmonary abnormalities seen on high resolution CT scanning.5
Although this illustrative case with a typical presentation of ACS had a favourable outcome, the question is raised of whether anything could have been done to avoid respiratory failure.
Pathophysiology of the acute chest syndrome in sickle cell disease
The genetic defect in sickle cell anaemia causes a substitution of valine for glycine in the β-globin subunit of haemoglobin to form HbS.6 HbS is less soluble than normal haemoglobin (HbA) when deoxygenated, as a result of which HbS polymerises within the cell. This stiffens the erythrocyte and changes it from its normal biconcave form to a sickle shaped cell. The sickle cell also loses the flexibility required to traverse capillary beds. Hypoxia also enhances adhesion of red cells to the vascular endothelium, a process mediated by interaction between very late activation antigen 4 (VLA-4) expressed on sickle cells and vascular cell adhesion molecule 1 (VCAM-1) on endothelial cells.7 As a consequence, the sickle cells occlude small and sometimes larger vessels causing vascular injury, especially to organs with sluggish circulation such as the spleen and bone marrow and in atelectatic areas of lung.
There are four precipitants of ACS: infection, atelectasis, fat embolism, and true thromboembolism (fig 1). Each may progress to a common final pathway of reduced ventilation with hypoxia and increased sickling. The most common finding in the lung during an acute painful crisis affecting the chest wall is atelectasis from a combination of poor chest expansion caused by painful rib and vertebral infarction and suppressed respiratory drive due to opiates. Atelectasis promotes sickling locally due to hypoxia causing inflammation, intravascular coagulation, and vascular obstruction with eventual micro-infarction. In adults, particularly, vaso-occlusion in the bone marrow causes marrow infarction and fat embolism. At post mortem examination many patients have bony spicules and marrow fat in the lung, and lipid laden macrophages can be found in bronchoalveolar lavage (BAL) fluid of patients with ACS.8 There is also increased circulating secretory phospholipase A2, a potent inflammatory mediator originating from the bone marrow, both in patients with ACS and those who are at risk of developing ACS, but not in those with uncomplicated vaso-occlusive crises.9,10
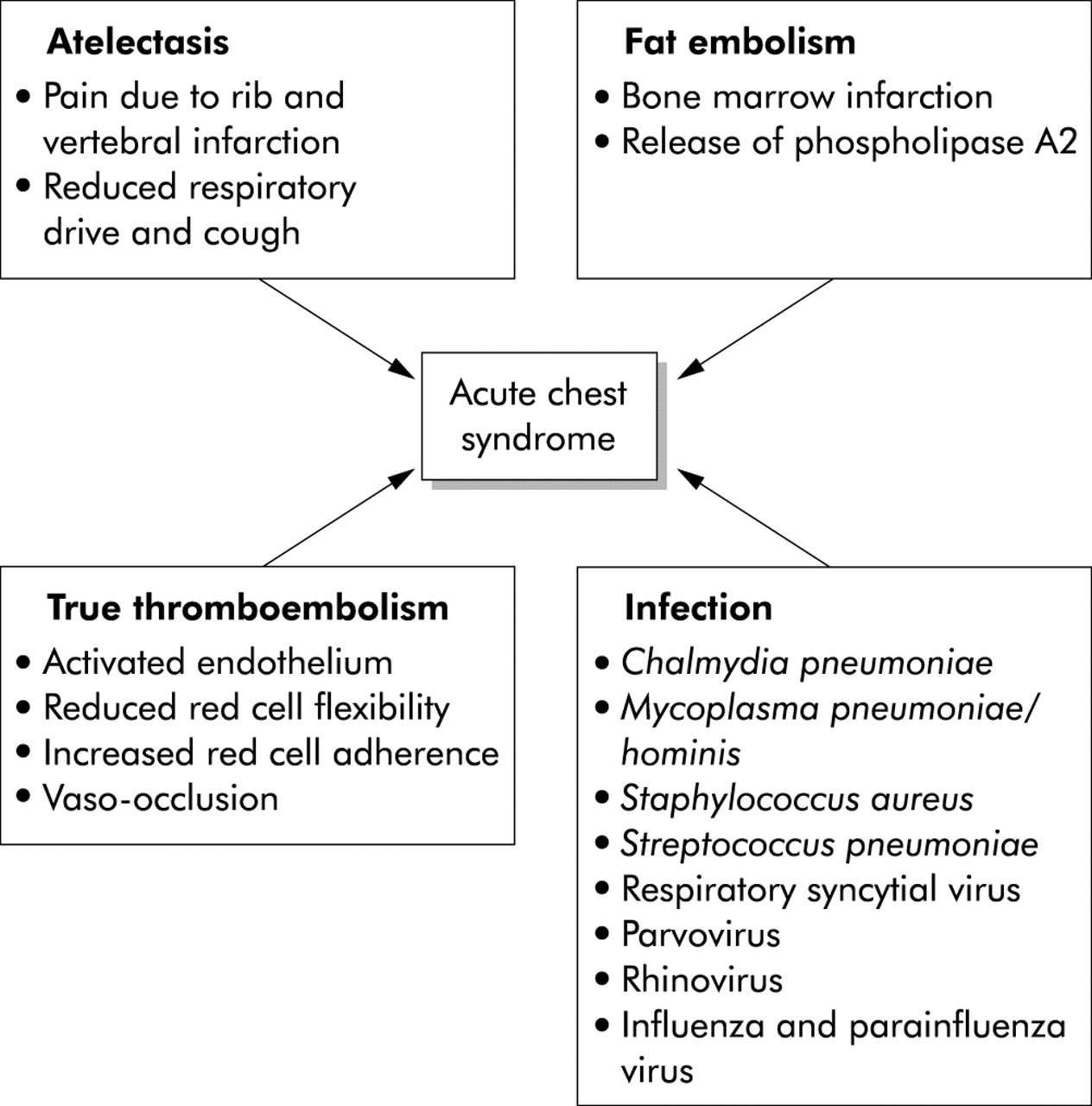
{kind=link}
Pathophysiology of the acute chest syndrome in sickle cell disease.
Nitric oxide (NO) is an endothelium derived vasodilator and a modulator of diverse inflammatory processes. Plasma concentrations of secretory VCAM-1 are raised in patients with ACS and are inversely related to plasma levels of NO metabolites.11 NO inhibits VCAM-1 expression, thus reduced endothelial synthesis of NO during vaso-occlusive crises and hypoxia may contribute to red cell adhesion within the lung. NO binds and increases HbS avidity for oxygen both in vitro and in vivo, and hence may reduce its tendency for polymerisation12 and thus has a potential therapeutic role in ACS.
NO is thought to play a significant role in the regulation of hypoxic pulmonary vasoconstriction. Exhaled concentrations of NO are directly related to the inhaled concentration of oxygen,13 suggesting that oxygen may be a rate limiting substrate for NO synthesis. Normal Hb has an affinity for NO that is 3000 times that of oxygen, so Hb may act as a “biological sink” for NO14 and rapid clearance of NO by Hb may contribute to hypoxic pulmonary vasoconstriction.15 However, in the presence of anaemia there may be failure of hypoxic vasoconstriction due to increased local levels of NO, despite reduced local production. Failure of hypoxic vasoconstriction worsens ventilation-perfusion matching, giving ideal conditions for further sickling and eventually ACS.
Hydroxyurea has made a big impact on the management of SCD. In a double blind, placebo controlled trial in 299 adults with sickle cell anaemia who had at least three painful episodes in the preceding year, hydroxyurea reduced the frequency of painful episodes, the incidence of ACS, and reduced the need for hospitalisation and transfusion.16 Hydroxyurea increases the concentration of fetal Hb (HbF) in erythrocytes, reducing the tendency for polymerisation of HbS.17,18 However, some clinical improvements are seen before the HbF concentration increases.19 In other studies hydroxyurea reduced adhesion of sickle cells to the vascular endothelium in vitro by reducing VCAM-1 expression.20 Hydroxyurea is also oxidised by heme groups to produce NO,21 and increased plasma NO metabolites can be detected during treatment.22 The beneficial effects of hydroxyurea may therefore be mediated via its properties as an NO donor as well as its effects on HbF.
Management of acute chest syndrome
Most adult patients are admitted with vaso-occlusive crises and develop ACS after a few days, whereas children are more likely to have a preceding febrile illness or infection.1,23 Once the process of lung injury has started it may be difficult to stop, and thus the aim of management should be to prevent ACS. Although the causes, clinical presentation, and outcomes of ACS have been well documented,1,2,23 less is known about its prevention or management.
General management
The most common cause for emergency admission of adults with SCD is a vaso-occlusive crisis. The mainstays of management are pain control, rehydration, oxygenation, and treatment of any identifiable precipitating cause. Common precipitants are infection, cold, stress, hypoxia and dehydration, but very often no obvious cause can be found. Infection—both bacterial and viral—is more common in children.
Adequate pain control usually requires initially high doses of opiates given by subcutaneous or intramuscular injection. The aim is to control pain rather than treating it as required. It is not uncommon for the doses required to suppress respiration and cough, causing atelectasis, retained secretions, and hypoxaemia. On the other hand, inadequate pain control may limit expansion and cough with the same consequences. In patients requiring opiates, intravenous rehydration is preferred aiming for 3–4 litres a day as the patient may not be able to drink adequate amounts owing to pain or drowsiness from excessive analgesia. Care should be taken not to cause fluid overload, especially in patients with impaired renal or cardiac function as a long term complication of SCD. Fluid overload will exacerbate pulmonary oedema associated with lung injury. Patients should be encouraged to drink freely.
There is a trend to give oxygen routinely to all patients with a vaso-occlusive crisis. However, if the oxygen saturations are normal (>97% on air), there is little to be gained from supplemental oxygen although it may reduce sickling in poorly ventilated areas of the lung. In patients with reduced respiratory drive or ventilation-perfusion mismatch, oxygen saturations may be reassuringly normal while the patient is on supplemental oxygen, masking the onset of acute lung injury. We recommend that pulse oximetry should be monitored regularly and all measurements should be performed after breathing room air for 10 minutes. If there is a fall of more than a few percent from normal values (patients with SCD may have low baseline levels due to chronic sickle lung disease), the cause should be sought and treated aggressively to prevent progression to ACS.
Specific management
Respiratory infection is a common precipitant of a sickle crisis but pathogens are rarely detected. In one study an identifiable pathogen was isolated in just over 30% of episodes,1 but this figure is dependent on how hard one looks. The two most common organisms were Chlamydia pneumoniae and Mycoplasma (mostly M pneumoniae and occasionally M hominis). Children suffered more infections with respiratory syncytial virus and parvovirus. Pneumococcus or Staphylococcus were less common, even though most patients are hyposplenic. However, in the reported study patients with chlamydial infections were less likely to be taking prophylactic antibiotics. Many of the cases of infection also had evidence of marrow infarction.1 Since atypical organisms predominate, a strong case can be made for treatment with macrolide antibiotics as the first line treatment when infection is thought to be the cause. However, caution should be exercised as the pattern of infectious agents in the UK may be different and further studies are required.
About 25% of patients with ACS wheeze and may respond to bronchodilators. Obstructive spirometry and small airways disease is not an uncommon finding in patients with SCD.1 Thus, in patients on mechanical ventilation, high airway pressure may be due to airway obstruction rather than reduced lung compliance, and gas trapping and intrinsic positive end expiratory pressure should be monitored. Routine use of incentive spirometry has recently been recommended to prevent atelectasis and ACS in patients with SCD admitted with chest or bone pain.24 In this randomised control study on 29 patients, 10 maximal inspirations with the incentive spirometer every 2 hours while the patient was awake significantly reduced the incidence of pulmonary complications. We prefer to use CPAP when the saturations fall below 93% on air or there is atelectasis on the chest radiograph. This is because pain and opiate sedation limits effort and compliance with active breathing techniques.
Blood transfusion and exchange transfusion are not required during uncomplicated painful episodes, but may be necessary when haemolysis is severe, if a large amount of blood is sequestrated in the spleen, or if there is an aplastic crisis caused by parvovirus infection. However, exchange transfusion can dramatically alter the course of ACS by replacing sickle cells with those containing HbA and by improving anaemia.25 In addition, transfusion also rapidly improves oxygenation, which suggests that anaemia may lead to increased local pulmonary NO accumulation increasing shunt by counteracting hypoxic vasoconstriction.26 Whether exchange transfusion is superior to simple transfusion is unclear, but in the US study of ACS both were effective with a low risk of alloimmunisation if phenotypically matched blood was used.1 Our practice is to use exchange transfusion aiming for HbS <20% and a total Hb of <14.5 g/dl. Where simple transfusion is used, care must be taken not to raise blood viscosity which promotes sickling, so the total final Hb should be <10.5 g/dl.
Inhaled NO is widely used to improve oxygenation in respiratory failure.27 Since NO downregulates endothelial adhesion molecule expression and increases the avidity of Hb for oxygen, reducing the tendency to sickling, there may be a specific role for NO supplementation in ACS. There have only been two reports of its use in three patients with ACS who have required mechanical ventilation.28,29 Inhaled NO (20–80 ppm) was used for 2–4 days while all three cases were aggressively treated with exchange transfusion, rehydration, and ventilatory support. All showed improvements in oxygenation and a reduction in pulmonary artery pressure on administration of NO, and all three patients survived. The use of inhaled NO in ACS has not been rigorously examined and as yet cannot be recommended.
CONCLUSIONS
New insights into the pathogenesis of ACS have highlighted potential therapeutic strategies—for example, involving NO. However, the focus of management of a patient admitted with a painful crisis must be to prevent progression to ACS. A multidisciplinary approach involving haematologists, chest physicians, sickle cell specialist nurses, and physiotherapists is therefore required with emphasis on adequate pain control, rehydration, adequate oxygenation, treatment of atelectasis, prompt treatment of infection, and the use of bronchodilators.
New insights into the pathophysiology of acute chest syndrome (ACS) have highlighted potential therapeutic strategies including the use of nitric oxide. A multidisciplinary approach to the management of patients admitted with a sickle cell crisis is needed to prevent progression to ACS and need for mechanical ventilation.