Article Text

Abstract
Background: The interactive roles of cytokines, endotoxins, superoxide (O2• − ) and nitric oxide (NO) in the pathogenesis of adult respiratory distress syndrome (ARDS) have not been fully elucidated. The effects of tumour necrosis factor-α (TNF-α), interleukin 1α (IL-1α), and lipopolysaccharide (LPS) and the role of NO and the endothelium in mediating O2• − formation were therefore investigated in intact porcine pulmonary arteries in vitro.
Methods: Intrapulmonary artery (PA) segments were obtained from White Landrace pigs (25–35 kg) and incubated with LPS, IL-1α, and TNF-α and O2• − release was measured by the superoxide dismutase (SOD) inhibitable reduction of ferricytochrome c. The source of O2• − formation was determined using a number of enzyme inhibitors. The role of NO was explored using NO synthase (NOS) inhibitors and the distribution of NOS isoforms and peroxynitrite (ONOO−, an index of NO–O2• − interactions) assessed by immunocytochemistry.
Results: LPS, IL-1α, and TNF-α promoted the formation of O2• − from PA compared with untreated controls in a time and dose dependent manner, an effect markedly enhanced by removal of the endothelium but completely inhibited by the NADPH oxidase inhibitor diphenylene iodonium chloride (DPI). l-NAME and the eNOS inhibitor N5-(1-iminoethyl)-ornithine (l-NIO) enhanced O2• − formation from PA (with endothelium) in response to IL-1α and TNF-α but had no effect on LPS mediated O2• − formation, whereas l-NAME and the iNOS inhibitor l-N6-(1-iminoethyl)-lysine-HCl (l-NIL) enhanced O2• − formation only in response to LPS.
Conclusions: LPS, IL-1α, and TNF-α promote O2• − formation through an upregulation of NADPH oxidase activity which is augmented by removal of the endothelium, as well as the inhibition of eNOS (in the case of cytokines) and iNOS (in the case of LPS). The concomitant expression of NOS isoforms (and NO formation) with that of NADPH oxidase may therefore constitute a protective system designed to remove O2• − through the formation of ONOO−. If this is so, the integrity of the endothelium may be axiomatic in the progression and severity of ARDS.
- adult respiratory distress syndrome
- nitric oxide
- superoxide
- cytokines
- endotoxin
Statistics from Altmetric.com
Oxidative stress plays a central role in the aetiology of adult respiratory distress syndrome (ARDS),1 a condition characterised by a time dependent worsening of intrapulmonary inflammation and hypertension.1–3 Principal among the reactive oxygen species (ROS) generated by oxidative stress is superoxide (O2• − ) which promotes vasoconstriction, damage to the endothelium, adhesion molecule expression, and thus perpetuation of inflammation.1,4 O2• − also reacts with nitric oxide (NO) to produce peroxynitrite (ONOO−) which, apart from reducing endogenous NO bioavailability, is inflammogenic in its own right.5 In turn, a reduction of NO availability promotes, not only vasoconstriction, but also the adhesion of leucocytes and platelets which release a battery of vasoconstrictors and cytokines.6 The negation of NO by O2• − therefore plays a key role in the pathogenesis of ARDS through the exacerbation of ongoing inflammatory cascades and the development of pulmonary hypertension.4 The importance of NO in ARDS has been confirmed by studies which have shown a beneficial effect of inhaled NO in ameliorating a reduction in leucocyte activity and oxidant stress.7
In ARDS there are several potential sources of O2• −. In the initial stages of the syndrome endotoxins activate neutrophils, monocytes and platelets and promote their adhesion to the pulmonary vessel walls.8 Neutrophils, through the respiratory burst reaction, then release large amounts of O2• − which reduces NO availability.9 Adherent neutrophils and monocytes also release tumour necrosing factor-α (TNF-α) and interleukins (ILs), the blood levels of which are markedly raised in patients with ARDS.10 In turn, lipopolysaccharide (LPS), TNF-α and ILs upregulate enzymes that generate O2• − in cultured vascular tissues (particularly NADPH oxidase11) and there is a causal relationship between increased O2• − formation and impaired vasodilation.12 It is therefore reasonable to suggest that endotoxins and cytokines augment O2• − in the pulmonary vasculature in ARDS.
The effect of LPS, IL-1α, and TNF-α on O2• − formation in whole porcine pulmonary artery (PA) segments was investigated in vitro, and the interactions between LPS, IL-1α, and TNF-α were examined. The effects were also studied over a 16 hour time course since ARDS is a dynamic time dependent process.1–3 The source of O2• − was determined with enzyme inhibitors of NADPH oxidase, xanthine oxidase, and mitochondrial respiration. Since little is known about the relative contributions of endothelial cells and vascular smooth muscle cells to overall O2• − formation in pulmonary arteries, O2• − formation was also measured in PA segments in which the endothelium had been removed. In addition, the role of inducible NO synthase (iNOS) and endothelial NO synthase (eNOS) in mediating overall O2• − production by PA was studied using specific inhibitors of these enzymes as well as immunocytochemistry.
METHODS
Dissection and incubation of pulmonary arteries
Lungs were obtained from White Landrace male pigs of 20–25 kg body weight. All animal experiments were conducted in accordance with the rules and regulations of Bristol University and the Home Office regarding the care and use of experimental animals. Pigs were anaesthetised with an intravenous injection of ketamine hydrochloride (10 mg/kg; Ketaset Injection, Fort Dodge Animal Health, Southampton, UK) and inhaled oxygenated halothane. The internal carotid artery was exposed, a cannula was placed in the carotid artery, and the animals were then exsanguinated. The chest was opened by median sternotomy and the lungs excised from the chest. Pulmonary arteries (PA) of 3–4 mm diameter (1st order) or 200–400 μm (4th order) were dissected from the lungs within 30 minutes and placed in Dulbecco’s Minimum Essential Medium supplemented with Glutamax-1, 100 U/ml penicillin, and 100 μg/ml streptomycin (DMEM; GibcoBRL, Paisley, UK). The arteries were cut into 2–3 mm2 segments for experimentation. In some studies the endothelium was removed by gently rubbing the luminal surface with a cotton wool bud.
The segments were then incubated for up to 16 hours in serum-free DMEM containing LPS (E coli 026:B6; Sigma, Poole, Dorset, UK), human recombinant IL-1α (R&D Systems, Abingdon, UK), and human recombinant TNF-α (R&D Systems) alone and in combination with each other. After incubation the segments were washed in Dulbecco’s phosphate buffered saline (PBS, GibcoBRL) and the formation of O2• − was measured.
Measurement of superoxide (O2• −)
The measurement of O2• − release by arterial segments was performed by detection of ferricytochrome c reduction.13 Following incubation, arterial segments were rinsed three times with PBS and equilibrated in DMEM without phenol red for 10 minutes at 37°C in a 95% air/5% CO2 incubator (Heraeus, Hera Cell, Kandro Laboratory Products, Germany). 20 μM horseradish cytochrome c (Sigma) with or without 500 U/ml copper-zinc superoxide dismutase (SOD; Sigma) was added to the segments and incubated at 37°C in a 95% air/5% CO2 incubator for 1 hour. The final volume of the reaction mixture was 0.5 ml/well. After 1 hour the reaction medium was removed and the maximum rate of reduction of cytochrome c was determined at 550 nm on a temperature controlled anthos Lucy 1 spectrometer (Lab-tech International, Ringmer, East Sussex, UK) and converted to nmol O2• − using ΔE550 nm = 21.1/mM/cm as the extinction coefficient for (reduced-oxidised) cytochrome c. The reduction of cytochrome c that was inhibitable with SOD reflected actual O2• − release. Segments were blotted, dried and weighed, and the data were expressed as nmol O2• − /mg tissue/h.
Effect of enzyme and NOS inhibitors on O2• − release
To determine the source of the O2• −, PA segments were preincubated with 10 μM diphenylene iodonium chloride (DPI, Sigma), an NADPH oxidase inhibitor; 10 μM rotenone (Sigma), an inhibitor of mitochondrial respiration; and 100 μM allopurinol (Sigma), an inhibitor of xanthine oxidase, for 2 hours before measurement of O2• −.
To study a possible role for NOS derived NO in modifying O2• − formation (O2• − + NO = ONOO−), the effect of NOS inhibitors was studied using (1) the non-specific NOS inhibitor l-nitroarginine methyl ester (l-NAME, 100 μM; Sigma), (2) the eNOS inhibitor N5-(1-iminoethyl)-ornithine (l-NIO, 10 μM; Sigma), and (3) the iNOS inhibitor, l-N6-(1-iminoethyl)-lysine-HCl (l-NIL, 10 μM; Sigma).14 The production of O2• − was measured by SOD inhibitable reduction of ferricytochrome c as above.
Immunocytochemistry of NOS and nitrated tyrosine
Immunocytochemical analysis of eNOS, iNOS, and nitrated tyrosine (NT) was carried out in selected samples following incubation for 16 hours with LPS (1 μg/ml), IL-1α, or TNF-α (both 10 ng/ml) which were then snap frozen in liquid nitrogen and stored at −80°C. Cryostat sections (8 μm) were prepared and fixed in acetone for 10 minutes. The endogenous peroxide activity was inhibited with 1.2% H2O2 in methanol for 30 minutes. Sections were then treated with horse (for NT staining) or goat serum (for eNOS and iNOS) diluted 1:3 with Tris-buffered saline (TBS; Sigma), pH 7.4, drained and incubated with monoclonal antibodies against eNOS, iNOS (Transduction Laboratories, Oxford, UK) and nitrated tyrosine (Upstate Biotechnology, Buckinghamshire, UK) at a dilution of 1:200 for iNOS and eNOS and 0.3 μg/ml for NT overnight at 4°C. After washing in TBS the sections were treated with either biotinylated goat anti-rabbit (for NT; 1:200 dilution) or biotinylated goat anti-mouse (for eNOS/iNOS; 1:200 dilution) for 1 hour at room temperature, washed and further treated for an hour with avidin-biotin-peroxidase complex (Dako Ltd, Ely, Cambridgeshire, UK) as described in the manufacturer’s manual. The bound antibody was visualised by addition of 0.05% diaminobenzidine (DAB, Dako) and 0.03% hydrogen peroxide in PBS, which formed an insoluble brown precipitate (positive staining). The nuclei were counterstained using Mayer’s haematoxylin, dehydrated, and mounted. Control slides (an irrelevant isotype matched antibody in place of the primary antibody) were prepared to test the specificity of staining.
Statistical analysis
Statistical analysis was carried out using Instat (Graphpad Software Inc, San Diego, USA). Before undertaking the study, power analysis was carried out from which it was determined that an n of 6 was required for statistical assurance. A Kolmogorov-Smirnov test showed that the data were normally distributed. The data are thus expressed as mean (SE), n=6. Analysis was performed using ANOVA and a post hoc unpaired two tailed Student’s t test with Bonferonni’s adjustment.
RESULTS
LPS, IL-1α, and TNF-α promoted the formation of SOD inhibitable O2• − from porcine PA segments compared with untreated controls in a time and concentration dependent manner (figs 1 and 2). Since maximal O2• − formation was observed after 16 hours incubation, this time point was used in all subsequent studies. Similarly, the concentrations at which optimal responses were obtained were used in subsequent experiments (10 ng /ml for both IL-1α and TNF-α, and 1 μg/ml for LPS). Removal of the endothelium enhanced the formation of O2• − from porcine PA segments compared with untreated controls in response to LPS, IL-1α, and TNF-α following incubation for 16 hours (fig 2). When comparing first order arterial segments (2–4 mm diameter) with fourth order arterial segments (200–300 μm diameter), with and without endothelium, O2• − formation and release in response to LPS, IL-1α and TNF-α was similar between the two different sized vessels, although the absolute release of O2• − was greater in the smaller vessels (fig 3).
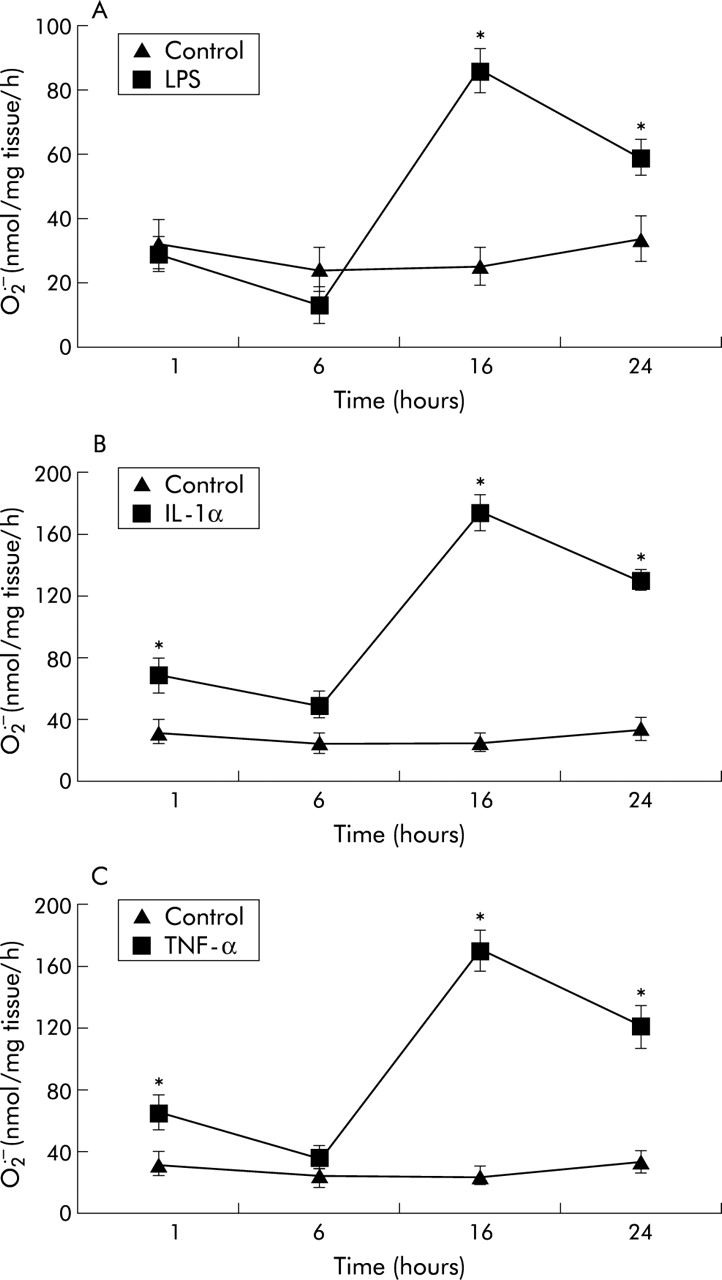
Time course of effects of (A) lipopolysaccharide (LPS; 1 μg/ml), (B) interleukin 1α (IL-1α; 10 ng/ml), and (C) tumour necrosis factor-α (TNF-α; 10 ng/ml) on SOD inhibitable superoxide (O2• − ) formation from porcine pulmonary artery segments compared with untreated controls. Each point is mean (SE); n=6. *p<0.001 treated v controls at each time point.

Effect of endothelium removal (–E) on SOD inhibitable O2• − formation from porcine pulmonary artery segments compared with segments with an intact endothelium (+E) in response to (A) lipopolysaccharide (LPS), (B) interleukin 1α (IL-1α), and (C) tumour necrosis factor α (TNF-α) following 16 hours incubation. Each point is mean (SE); n=6. *p<0.001 –E v +E at each concentration of cytokine or LPS; $p<0.001 responses of LPS or cytokines v zero (–E only); #p<0.001 responses of LPS or cytokines v zero (+E only).

SOD inhibitable O2• − formation by fourth order (200 μm diameter) porcine pulmonary artery segments with (+E) and without (–E) endothelium in response to lipopolysaccharide (LPS, 1 μg/ml), interleukin 1α (IL-1α, 10 ng/ml), and tumour necrosis factor α (TNF-α, 10 ng/ml) following 16 hours incubation. Values are mean (SE); n=6. *p<0.001 –E v +E for each treatment.
DPI, an inhibitor of NADPH oxidase (but not rotenone) inhibited the formation O2• − formation and release from porcine PA segments (with and without endothelium) in response to LPS, IL-1α, and TNF-α (fig 4). Allopurinol had a slight but still significant effect on O2• − formation induced with LPS and TNF-α but had no effect on IL-1α induced effects (fig 4).
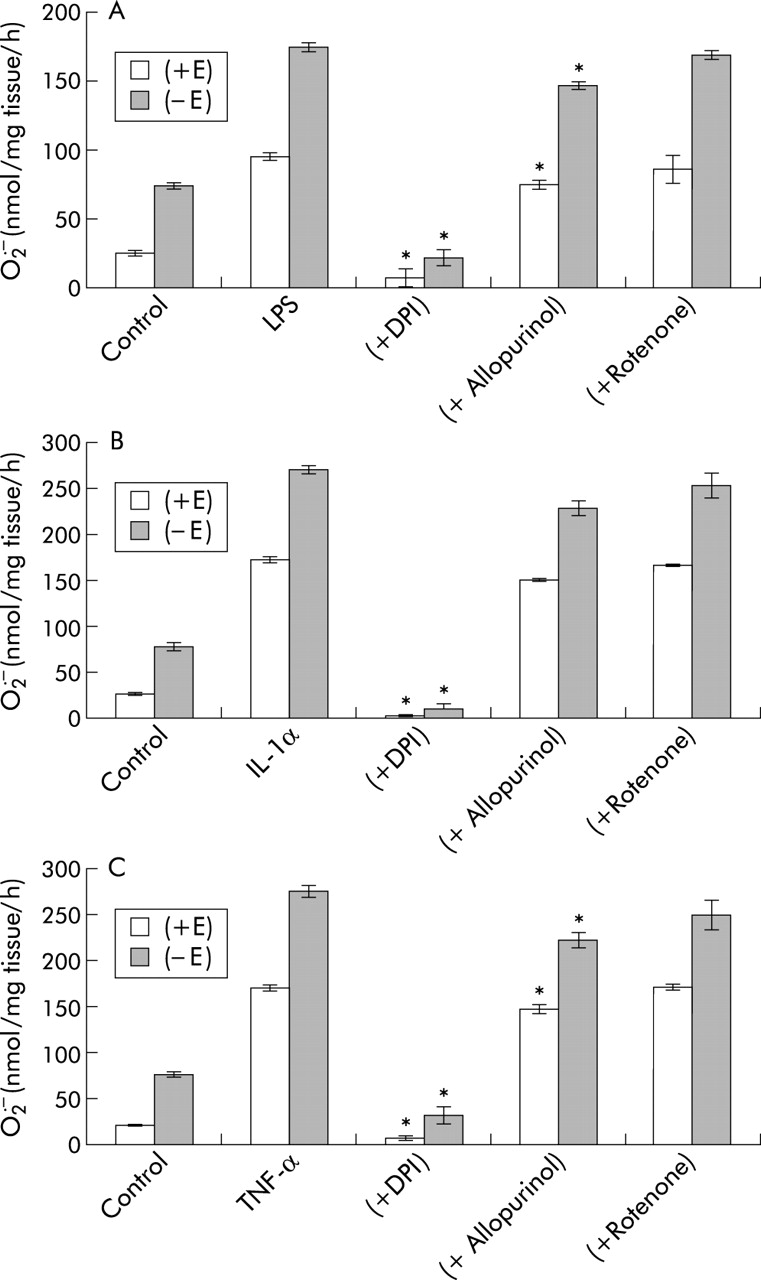
Effect of diphenylene iodonium chloride (DPI; 10 μM), allopurinol (100 μM), and rotenone (10 μM) on SOD inhibitable O2• − formation by porcine pulmonary artery segments with (+E) and without (–E) endothelium in response to (A) LPS (1 μg/ml), (B) IL-1α (10 ng/ml), and (C) TNF-α (10 ng/ml) following 16 hours incubation. Values are mean (SE); n=6. *p<0.01 significantly inhibited compared with LPS or cytokine treated segments.
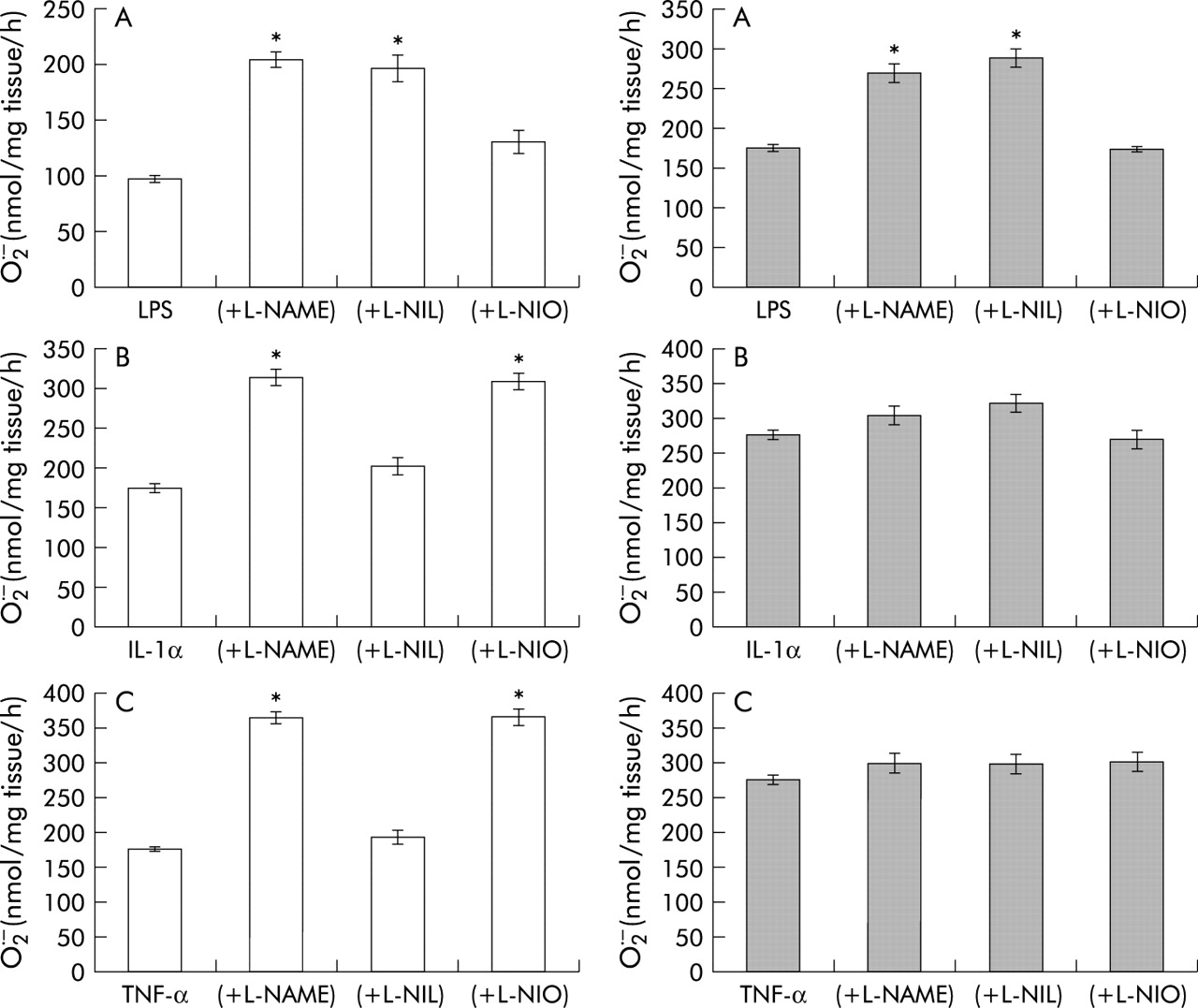
The non-specific NOS inhibitor l-NAME enhanced O2• − formation and release from porcine PA segments with and without endothelium in response to IL-1α and TNF-α, but only when the endothelium was present in response to LPS (fig 5). The eNOS inhibitor l-NIO enhanced O2• − formation from porcine PA segments with endothelium only in response to IL-1α and TNF-α and had a lesser effect on LPS mediated O2• − formation (fig 5). The iNOS inhibitor, on the other hand, enhanced O2• − formation from porcine PA segments with endothelium only in response to LPS, but had no effect on IL-1α and TNF-α mediated O2• − formation (fig 5).

Effect of the NOS inhibitors l-nitroarginine methyl ester (l-NAME, 100 μM), N5-(1-iminoethyl)-ornithine (l-NIO, 10 μM), and l-N6-(1-iminoethyl)-lysine-HCl (l-NIL, 10 μM) on SOD inhibitable O2• −- from porcine pulmonary artery segments in response to (A) lipopolysaccharide (LPS, 1 μg/ml), (B) interleukin 1α (IL-1α, 10 ng/ml), and (C) tumour necrosis factor α (TNF-α, 10 ng/ml) following 16 hours incubation in intact arteries (left) and without endothelium (right). Values are mean (SE); n=6. *p<0.001 v LPS or cytokine treated only.
Following incubation for 16 hours the immunoreactive distribution of eNOS, iNOS, and nitrated tyrosine (index of ONOO− formation) in response LPS, IL-1α, and TNF-α was assessed. Nitrated tyrosine was located principally in the endothelium (fig 6A–E), indicating that both NO and O2• − are present at high concentrations in this region. Similarly, eNOS was located in the endothelium and was markedly upregulated by IL-1α and TNF-α and, to a lesser extent, by LPS (fig 6F–J), confirming the effects of the NOS inhibitors. In contrast, iNOS was upregulated only in response to LPS and was located ubiquitously through the arterial segment, and not to IL-1α or TNF-α (fig 6K–O), again confirming the observations with the NOS inhibitors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Distribution of immunoreactive nitrated tyrosine and expression of eNOS and iNOS in pig pulmonary arteries. Freshly prepared pulmonary arterial segments were treated with vehicle (A, F, K), 1 μg/ml lipopolysaccharide (B, G, L), 10 ng/ml IL-1α (C, H, M), 10 ng/ml TNF-α (D, I, N) or with control IgGs (E, J, O). Frozen sections of these segments were then immunostained for either nitrated tyrosine (A–E), eNOS (F–J) or iNOS (K–O). Dark brown staining represents positive staining. The endothelium is indicated by arrowheads.
DISCUSSION
This study shows that TNF-α, IL-1α, and LPS promote the formation of O2• − in PA segments (with and without intact endothelium) in a time dependent manner and at concentrations that have been reported to appear in the blood of patients with ARDS.10 These responses were not confined to large pulmonary arteries but were seen also in arterial microvessels, indicating that these effects occur ubiquitously throughout the pulmonary vasculature. Furthermore, DPI completely inhibited the generation of O2• − in response to TNF-α, IL-1α and LPS, indicating that an increase in NADPH oxidase activity mediates these effects. The slight reduction in O2• − formation by allopurinol suggests that xanthine oxidase may also be upregulated.
These data are in agreement with previous observations. IL–1β has been shown to induce O2• − production from cultured rat pulmonary microvascular smooth muscle cells, human internal mammary artery smooth muscle cells, and human cord vein endothelial cells.15,16 TNF-α activated O2• − producing NADH oxidase in cultured rat aortic smooth muscle cells in a time and dose dependent manner.17 Increased O2• − has also been reported in whole lungs from guinea pigs following the injection of TNF-α.18 LPS challenge also increases lung O2• − generation in anaesthetised rats, which was found to coincide with the alterations in cardiovascular function measured in these animals.19 Clinically, it has long been recognised that intrapulmonary oxidative stress is present in patients with ARDS.20,21
Removal of the endothelium resulted in a marked increase in O2• − release from PA segments both before and after incubation with LPS, TNF-α, and IL-1α, indicating that the endothelium of PAs possesses a system for removing O2• − radicals. Although there are reports that the endothelium produces O2• −,22 our results show that the endothelium quenches the output of O2• − by the arterial segment as a whole. Since the endothelium is a principal source of NO, the reaction of O2• − with NO may account for the removal of O2• − by the endothelium and, as such, was investigated using NO synthase (NOS) inhibitors. The non-specific NOS inhibitor l-NAME mimicked the effect of endothelium removal—that is, O2• − release was increased by l-NAME in segments with intact endothelium—indicating that NO removes O2• − from the system through the reaction: NO + O2• − → ONOO−, thereby reducing that which reacts with ferricytochrome c. Indeed, immunocytochemical analysis clearly showed a marked increase in NT (an index of ONOO− formation) in the endothelial region of PA segments after incubation with cytokines and LPS, supporting this proposal.
To differentiate between the roles of eNOS and iNOS in reducing O2• − formation in intact PAs, the effect of two inhibitors of these isoforms were investigated. The eNOS inhibitor l-NIO, but not the iNOS inhibitor l-NIL, enhanced O2• − formation in response to IL-1α and TNF-α and, to a lesser extent, to LPS. Immunocytochemical analysis confirmed that eNOS (but not iNOS) is upregulated in the endothelium of the arterial segments in response to these cytokines and that LPS had no effect. It therefore appears that cytokines (principally) elicit a simultaneous upregulation of both smooth muscle NADPH oxidase and endothelial eNOS which negates the increased formation of O2• −, thereby protecting the vessel against oxidative damage. In turn, loss of the endothelium would render the PA susceptible to the pathogenic impact of cytokine induced O2• − formation. These data are also in agreement with those of Bhagat et al23 who concluded that IL-1β upregulates eNOS but not iNOS in venous tissue of human subjects in vivo.
In contrast, the iNOS inhibitor l-NIL enhanced O2• − formation in response to LPS but had no effect on IL-1α or TNF-α mediated O2• − formation. Immunocytochemical analysis again confirmed an upregulation of iNOS in response to LPS but no effect of cytokines on this isoform. It therefore appears that LPS has little effect on the upregulation of eNOS but influences the expression of iNOS at the level of the vascular smooth muscle. Nevertheless, NO produced by iNOS appears also to have a protective effect against LPS induced O2• − formation. Other studies have shown that LPS promotes the upregulation of iNOS while suppressing the expression of eNOS.24,25 Additionally, as these data indicate, the excess formation of ONOO− may contribute to the pathophysiology of ARDS directly since ONOO− has been shown to exert pathological effects including endothelial cell apoptosis and the expression of adhesion molecules.26,27 Clinically, raised levels of NO metabolites are present in high levels in bronchoalveolar lavage (BAL) fluid from patients with ARDS, which is consistent with an upregulation of eNOS and iNOS in intrapulmonary tissues in this condition.28 More crucially, increased levels of nitrotyrosine residues (index of ONOO−) have been reported in BAL fluid from patients with acute lung injury,29,30 consolidating the hypothesis that upregulation of both NO and O2• − generating enzymes occurs in ARDS by the mechanisms proposed in this study.
In conclusion, the present study shows that LPS and two major cytokines additively promote the formation of O2• − by pig pulmonary arteries in vitro, principally through an upregulation of NADPH oxidase. In turn, this O2• − would perpetuate and augment inflammation through the induction of adhesion molecule expression, vasoconstriction, and the negation of NO bioavailability. In this situation the endothelium reduces LPS and cytokine induced O2• − generation, a mechanism that appears to be mediated through NO derived from eNOS (in the case of cytokines) and iNOS (in the case of LPS). The concomitant expression of these NOS isoforms with that of NADPH oxidase may therefore constitute an important protection system against oxidative stress. However, at some point in the sequelae of ARDS this defence system may be overwhelmed, rendering the vasculature susceptible to oxidative stress. The integrity of the endothelium may thus be axiomatic in the progression and severity of ARDS. A reduction in oxidative stress coupled with the delivery of NO would therefore seem to be a potentially effective strategy for treating ARDS.
Acknowledgments
This research was supported by grants from the British Heart Foundation and the British Journal of Anaesthesia.