Article Text

Abstract
Background: Sarcoidosis is a systemic granulomatous disorder of unknown origin. Lymphocytic inflammation is dominated by expression of Th1 type cytokines such as tumour necrosis factor α (TNFα). Interleukin 13 (IL-13) is a Th2 cytokine which is expressed by CD4+ T cells and has been shown to suppress TNFα in human blood monocytes. The role of IL-13 as a possible anti-inflammatory cytokine in sarcoidosis was investigated.
Methods: mRNA expression of IL-13, IL-4, IL-10, and TNFα in bronchoalveolar lavage (BAL) fluid cells and peripheral mononuclear blood cells (PBM) of 18 patients with sarcoidosis and nine healthy controls was assessed using RT-PCR. In addition, IL-13 protein levels in BAL cell culture supernatants from 12 patients and all controls were measured and immunocytochemistry of IL-13 protein was performed in BAL fluid cells of eight patients. TNFα concentrations were measured with and without stimulation with recombinant human (rh) IL-13, rhIL-10, and lipopolysaccharide (LPS).
Results: IL-13 mRNA expression was significantly increased in BAL cells and PBM of patients compared with controls (p<0.05). No significant difference was found in IL-4 mRNA or IL-10 mRNA expression in BAL fluid cells or PBM between the two groups. TNFα mRNA expression was significantly higher in BAL fluid cells of patients than controls (p<0.05). IL-13 protein levels in BAL cell culture supernatants were slightly raised in half the patients investigated but in only two controls. Immunocytochemistry detected IL-13 protein in alveolar macrophages of patients. IL-13 led to decreased TNFα concentrations (p<0.05).
Conclusions: IL-13 expression is increased in BAL cells and PBM in sarcoidosis and IL-13 is secreted from BAL cells. Alveolar macrophages may be the cellular source. These data suggest that IL-13 might have an anti-inflammatory effect by acting on TNFα.
- interleukin-13
- sarcoidosis
- tumour necrosis factor α
Statistics from Altmetric.com
Sarcoidosis is a granulomatous systemic disorder which often involves the lungs and thoracic lymph nodes.1 Although the aetiology of this disease is unknown, recent investigations have given some insight into its pathophysiology. Current concepts include alveolar macrophages, T helper (CD4+) cells, and a cytokine network which act together to cause a compartmentalised inflammatory process. Spontaneous release of Th1 cytokines such as interferon (IFN)-γ and interleukin (IL)-2 by T cells of the bronchoalveolar lavage (BAL) fluid has been shown.2,3 Moller et al found higher expression of IL-12p40 mRNA in BAL cells of patients with sarcoidosis than normal controls.4 This increased level of IL-12 upregulates the development of Th1 cells and amplifies the release of Th1 cytokines. Alveolar macrophages have been shown to release a number of cytokines such as tumour necrosis factor α (TNFα) and IL-6.5–7 TNFα, in particular, is released in high concentrations and might be responsible for the chronic fatigue often seen in sarcoidosis patients, although a corresponding cachectin effect is obviously absent.8 More than 80% of patients do not suffer from serious disease and clinical symptoms are mild or exhibit spontaneous remission. It may therefore be possible that TNFα binding or neutralising proteins or counteracting cytokines are simultaneously released.9 Increased levels of soluble TNF receptors (TNF-R) have been found in the plasma and BAL fluid of subjects with sarcoidosis.10,11 Armstrong et al12 found that soluble TNF-R may inhibit TNFα bioactivity in the lungs of sarcoidosis patients.
Anti-inflammatory cytokines have not been well characterised. Information is available on transforming growth factor-β (TGFβ) and IL-10. Increased levels of TGFβ have been reported in the BAL fluid of patients with sarcoidosis,13 but a recent study also reported increased TGFβ levels in the BAL fluid of patients with altered lung function.14 The role of TGFβ therefore still needs to be defined. Enhanced secretion of IL-10 has been observed from alveolar macrophages but not from BAL fluid cells of patients with sarcoidosis.13,15
IL-13 is a recently described cytokine which shares some effects with IL-4.16,17 It is mainly produced by CD4+ T helper cells18,19 and has been shown to be involved in the pathogenesis of bronchial asthma.20 A previous study found that IL-13 could suppress TNFα secretion by human blood monocytes in healthy subjects,21 so it might have some anti-inflammatory effects or influence the Th1 immune response in sarcoidosis.
As information about IL-13 in sarcoidosis is sparse, mRNA expression in BAL fluid cells and peripheral blood mononuclear cells (PBM) in patients with sarcoidosis and in healthy controls was investigated. The mRNA expression of IL-4 and IL-10 was also determined to evaluate whether these cytokines might contribute to the suppression of proinflammatory reactions in sarcoidosis. The expression of TNFα mRNA was also investigated. IL-13 protein levels were measured in BAL cell culture supernatants. Immunocytochemistry was performed for identification of the cellular source of IL-13 in BAL cells. To investigate the effect of IL-13 on TNFα secretion, BAL cell cultures of patients were incubated with either recombinant human IL-13 (rhIL-13) or recombinant human IL-10 (rhIL-10).
METHODS
Patients and controls
Eighteen patients (10 men) of mean age 40 years (range 23–57) with sarcoidosis were investigated. The diagnosis of sarcoidosis was established by clinical signs, histological findings, and chest radiography. The CD4/CD8 ratio was determined by immunocytochemistry as described below. The mean serum angiotensin converting enzyme level was 60.6 (9.7) U/l. Six patients were classified as stage I, nine as stage II, and three as stage III (table 1). Six patients were treated with corticosteroids. No patient had a history or clinical symptoms of allergic disease.
Characteristics of study subjects
Nine healthy volunteers (five men) with a mean age of 27.5 years (range 21–31) served as controls. They had no history or clinical signs of lung or allergic disease.
Pulmonary function was measured in all groups by spirometry, body plethysmography, and gas exchange status from capillary blood (BodyScope, Medizin Elektronic GmbH, Germany). The forced expiratory volume in 1 second (FEV1), vital capacity (VC), and carbon monoxide transfer factor (Tlco) were used to describe the status of the lungs in patients and FEV1 and VC were used in controls. Tlco was determined in 11 patients.
Cell isolation and RNA preparation
BAL cells were isolated by centrifugation and washed once with phosphate buffered saline (PBS) containing 2% fetal calf serum (FCS; Seromed, Berlin, Germany). PBM were isolated by centrifugation over Ficoll-Paque (Pharmacia, Uppsala, Sweden) and washed twice with PBS containing 2% FCS. RNAzol (Wak, Bad Soden, Germany) was added and samples (80 000 cells each) were stored at −20°C until needed. RNA was extracted by treatment with chloroform and precipitated with isopropanol. The concentration of RNA in each sample was adjusted to 0.15 μg/μl and the samples were stored at −20°C.
RT-PCR
RNA was reverse transcribed using 2 μl MgCl2 (25 mM), 1 μl 10 × PCR buffer, 1 μl 10 mM of each deoxynucleotide triphosphate (dNTP), 0.5 μl Oligod T (50 μM), 0.5 μl RNAse inhibitor (20 U/μl), and 0.5 μl reverse transcriptase (50 U/μl; Perkin Elmer Biosystems, Roche, Branchburg, USA). The mixture was incubated at 42°C for 30 minutes and at 90°C for 5 minute. Samples were stored at −20°C until amplification.
The resultant cDNA was amplified in a thermal cycler (Hybaid, Teddington, UK) with a final volume of 100 μl containing 10 μl cDNA (IL-13, IL-4, TNFα, or IL-10) or 5 μl cDNA (β-actin), 10 μl or 15 μl dilution buffer, 8 μl MgCl2 (12 mM), 8 μl 10 × PCR buffer, 55.5 μl H2O with DEPC, 0.5 μl recombinant Taq DNA polymerase (5 U/μl; Perkin Elmer Biosystems, Roche, Branchburg, USA), and 2.0 μl 15 mmol of each primer. The oligonucleotide primers for PCR were based on published mRNA sequences. β-actin served as control. The human β-actin primers were 5′-GTG GGG CGC CCC AGG CAC CA-3′ for the upstream primer and 5′-CTC CTT AAT GTC ACG CAC GAT TTC-3′ for the downstream primer; IL-13 used 5′-GAG TGT GTT TGT CAC CGT TG-3′ for the upstream primer and 5′-TAC TCG TTG GTC GAG AGC TG-3′ for the downstream primer; IL-4 primers were 5′-ATT TCT CTC TCA TGA TCG TC-3′ for the upstream primer and 5′-GGA CAC AAG TGC GAT ATC ACC-3′ for the downstream primer; IL-10 primers used 5′-ATG CCC CAA GCT GAG AAC CAA GAC CCA-3′ for the upstream primer and 5′-TCT CAA GGG GCT GGG TCA GCT ATC CCA-3′ for the downstream primer; and TNFα primers were 5′-CAG AGG GAA GAG TTC CCC AG-3′ for the upstream primer and 5′-CCT TGG TCT GGT AGG AGA CG-3′ for the downstream primer. PCR amplification was performed for 36 cycles (1 min at 94°C, 1 min at 60°C, 40 seconds at 72°C) for β-actin, for 35 cycles (45 seconds at 95°C, 45 seconds at 60°C, 90 seconds at 72°C) for IL-13, for 38 cycles (1 min at 94°C, 1 min at 60°C, 1 min at 72°C) for IL-4, for 35 cycles (1 min at 94°C, 1 min at 59°C, 40 seconds at 72°C) for IL-10, and for 40 cycles (1 min at 94°C, 1 min at 60°C, 40 seconds at 72°C) for TNFα.
Identification of PCR products
PCR products were analysed by electrophoresis on a 2% agarose gel containing ethidium bromide and visualised by UV light. The sizes of the PCR products were compared with the expected PCR product length and molecular weight markers (Boehringer Mannheim, Germany).
Densitometric and semiquantitative PCR analysis
Densitometric analysis was performed by the Eagle Eye II Still Video System (Stratagene, La Jolla, USA). All cytokine signals produced by RT-PCR were within the linear range of the assay. The expression was standardised to that of β-actin expression from the same reverse transcribed cytokine (IL-13, IL-4, IL-10, TNFα) mRNA sample. Ratios of IL-13:β-actin, IL-4:β-actin, IL-10:β-actin, and TNFα:β-actin were calculated for semiquantitative RNA expression analysis as previously described.22
Immunocytochemistry (ICC)
BAL cells were fixed on glass slides and permeabilised with acetone for 4 minutes. Anti-human CD4 (Dako, Denmark) and anti-human CD8 (Dako, Denmark) mouse antibodies for each patient sample were added for 1 hour. Anti-human IL-13 mouse antibody (R&D Systems, Minneapolis, USA) or specific isotype control were added to samples of eight patients. Slides were then washed with Tris buffer containing 10% fetal calf serum (Tris+FCS) and anti-mouse rabbit antibody and anti-rabbit pig antibody were added for 30 minutes. After washing with Tris+FCS for 5 minutes the slides were incubated with a soluble complex of alkaline phosphatase and antibody to alkaline phosphatase (APAAP) for 30 minutes and washed for 5 minutes with Tris+FCS. This procedure was repeated once for enhancement. Fast red substrate containing 2 mg naphthol, 200 μl dimethylformamide (DMF), 2.4 mg levamisole, and 10 mg fast red in 9.8 ml 0.1 M Tris buffer was added to the cells for a maximum of 20 minutes (staining intensity was visually controlled). After washing with water for 5 minutes the cells were counterstained with haemalaun for 2 minutes and washed for a further 10 minutes.
The CD4/CD8 ratio was determined by counting the number of positive cells among 200 lymphocytes from each staining. For IL-13, ICC isotype controls were used to determine the intensity of staining; 200 alveolar macrophages were counted in each sample and the percentage of positive cells was calculated. All slides were counted by two experienced observers. The interobserver difference was below 5%.
BAL cell cultures
BAL cells were cultured in RPMI 1640 medium containing 10% FCS, 2% l-glutamine, and 1% penicillin/streptomycin (Biochrom KG, Berlin, Germany). The concentration was 1000 cells/μl. After incubation for 18 hours the samples were centrifuged to obtain supernatants which were stored at −20°C until needed. Viability, as determined by trypan blue exclusion, was usually >85%. BAL cell cultures from 12 patients and all controls were incubated for measurement of IL-13. The cell cultures from the 12 patients were incubated both without and with recombinant human IL-13 (rhIL-13; R&D Systems, Minneapolis, USA) at a concentration of 5 ng/ml as indicated by the manufacturer. Cell cultures from six patients were also stimulated with lipopolysaccharide (LPS) at a concentration of 1.0 μg/ml, both with and without the addition of rhIL-13. The BAL cell cultures from the other six patients were incubated without and with recombinant human IL-10 (rhIL-10) at a concentration of 0.4 μg/ml as indicated by the manufacturer (Strathmann Biotech GmbH, Hannover, Germany).
Enzyme-linked immunosorbent assays (ELISA)
An IL-13 ELISA (R&D Systems) was performed to measure IL-13 protein levels in BAL cell culture supernatants after 18 hours of incubation in 12 patients and all controls. A TNFα ELISA (R&D Systems) was used to determine the concentration of TNFα in cell culture supernatants of patients with and without the addition of rhIL-13 (n=12) or rhIL-10 (n=6). Concentrations of IL-13 and TNFα were determined by comparison with recombinant standards run parallel with each batch of assay.
Analysis of data
A Mann-Whitney test was used to compare cytokine expression in patients and controls. To compare TNFα protein levels in cell culture supernatants without and with stimulation by LPS, rhIL-13 or rhIL-10, a paired t test was used. A p value of <0.05 was considered statistically significant.
RESULTS
BAL and PBM total and differential cell counts, CD4/CD8 ratio, FEV1, VC and Tlco
The results of BAL and PBM total cell counts as well as differential cell counts in both groups are summarised in table 2. The mean (SE) CD4/CD8 ratio as determined by ICC in the patient group was 3.8 (0.7). VC was 89.1 (3.5)% predicted in the patient group and 95.5 (1.8)% predicted in the control group; FEV1 was 81.6 (5.1)% predicted in the patients and 100.3 (2.7)% predicted in the controls; and Tlco was 80.8 (3.3)% predicted in the patient group (table 1).
BAL fluid and PBM cell counts of patients and control subjects
Expression of IL-13, IL-4, IL-10, and TNFα mRNA
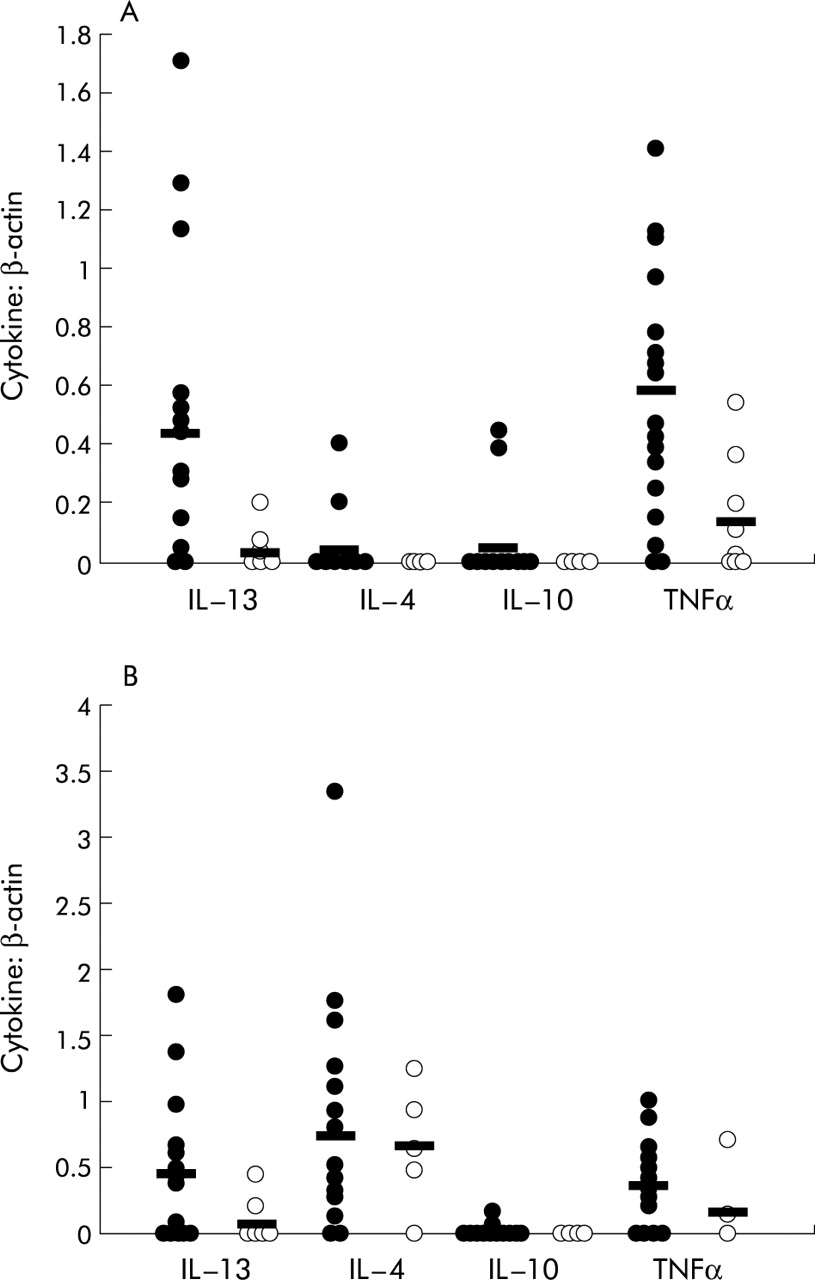
All but three patients showed IL-13 mRNA expression in BAL fluid cells with a mean (SE) IL-13:β-actin ratio of 0.44 (0.13), whereas BAL fluid cells of only three of the control group expressed IL-13 (mean value 0.03 (0.13), fig 1). In PBM seven patients did not express IL-13 while the other patients showed strong expression. The resulting mean IL-13:β-actin ratios was 0.45 (0.10). IL-13 expression was detectable in PBM of only three controls (mean 0.07 (0.05), fig 1). The differences between the two groups in both BAL fluid cells and PBM were statistically significant (p<0.05; table 3, fig 2) Further analysis of IL-13 mRNA expression in BAL fluid cells of patients was performed by comparing patients with stage I disease with those with stage II/III disease. IL-13 mRNA expression in the group with stage I disease was significantly increased compared with the group with stage II/III disease (0.76 (0.24) versus 0.27 (0.09); p<0.05, fig 3). There was no significant difference in IL-13 mRNA expression in BAL fluid cells between patients treated with corticosteroids and untreated patients.
Expression of IL-13, IL-4, IL-10, and TNFα in BAL fluid cells and PBM of patients and controls

IL-13 mRNA expression in BAL fluid cells and peripheral blood mononuclear cells (PBM) of patients and controls. Lanes 1–8: β-actin expression in BAL cells (lanes 1–2) and PBM of patients (lanes 3–4) and BAL cells (lanes 5–6) and PBM (lanes 7–8) of controls. Lanes 9–16: IL-13 expression could be detected in BAL cells (lanes 9–10) and PBM (lanes 11–12) of patients but not in BAL cells (lanes 13–14) or PBM (lanes 15–16) of controls. The length of the IL-13 transcripts was 275 bp as expected. M=molecular weight marker.

Semiquantitative PCR analysis of mRNA expression of (A) BAL fluid cells and (B) peripheral blood mononuclear cells (PBM) in patients (•) and controls (ø). Cytokine expression of IL-13, IL-4, IL-10, and TNFα was normalised to β-actin and ratios were calculated. Mean values are shown by horizontal lines. The difference in IL-13 expression in BAL fluid cells and PBM between patients and controls was statistically significant; the differences in IL-4 and IL-10 expression were not significant.

IL-13 mRNA expression in sarcoidosis patients according to stage of disease. Stages II and III were combined. Circles indicate values of semiquantitative PCR analysis. Mean values are shown by horizontal lines.
IL-4 mRNA was expressed in the BAL fluid cells of only three patients. In the other patients no IL-4 mRNA could be detected. The mean IL-4:β-actin ratio for the whole group was 0.04 (0.03). IL-4 mRNA was not expressed in the BAL fluid cells of the five controls in which it was measured. PBM of all but three patients expressed IL-4 mRNA with a mean IL-4:β-actin ratio of 0.75 (0.13). In controls IL-4 was strongly expressed in PBM with a mean IL-4:β-actin ratio of 0.66 (0.21), n=5. Nevertheless, the differences between BAL fluid cells and PBM of patients and controls were not statistically significant (table 3, fig 2).
IL-10 mRNA expression could only be detected in BAL fluid cells of two patients with IL-10:β-actin ratios of 0.45 and 0.39. In the control group IL-10 was also expressed only in BAL fluid cells of two volunteers with lower IL-10:β-actin ratios of 0.02 and 0.09. IL-10 was expressed in PBM of three patients with values of 0.12, 0.05, and 0.16, and could be detected in PBM of the six controls tested in only very small amounts (<0.01, table 3, fig 2).
TNFα mRNA was expressed in BAL fluid cells of all but one patient with a mean TNFα:β-actin ratio of 0.58 (0.09). In the control group TNFα mRNA expression was detected in about half the BAL fluid cells (5 of 9) with a mean value of 0.14 (0.07). This difference was statistically significant (p<0.05). TNFα mRNA was expressed in the PBM of 11 patients with a mean value of 0.35 (0.08). In the control group TNFα mRNA was expressed in the PBM of two of the five tested with a mean value of 0.17 (0.14). This difference was not statistically significant (table 3, fig 2).
IL-13 levels in BAL cell culture supernatants
IL-13 protein concentrations in BAL cell cultures were slightly increased in six of the 12 patients tested (range 6.01–27.70 pg/ml). Only two controls had increased IL-13 levels (12.97 pg/ml and 18.75 pg/ml). Median values were 3.01 pg/ml in the patients and 0.00 pg/ml in the controls. This difference was not statistically significant.
IL-13 protein in BAL fluid cells of patients
Immunocytochemistry of IL-13 protein in BAL fluid cells of eight patients showed positive staining of alveolar macrophages with a mean (SE) of 30.5 (8.6)% positive cells. Only a few lymphocytes were stained, but the intensity was much lower than for the alveolar macrophages (fig 4).

Immunocytochemistry of IL-13 protein in BAL fluid cells of patients with sarcoidosis (original magnification ×200). Alveolar macrophages stained positively for IL-13 whereas lymphocytes did not.
Effect of stimulation with LPS/rhIL-13/rhIL-10 on TNFα secretion
After 18 hours of incubation with rhIL-13 or rhIL-10 the TNFα protein concentration was decreased in BAL cell culture supernatants compared with unstimulated cells. The mean TNFα levels were 1068.12 (228.43) pg/ml in unstimulated cells, 909.43 (196.15) pg/ml in cells stimulated with rhIL-13 (n=12; p<0.05), and 959.92 (231.33) pg/ml in those stimulated with rhIL-10 (n=6; p<0.05). LPS stimulation significantly increased TNFα levels compared with unstimulated cells (1713.24 (272.53) pg/ml; n=6; p<0.05). The addition of rhIL-13 significantly reduced TNFα concentrations in LPS stimulated BAL cell cultures (1202.86 (41.89) pg/ml; n=6; p<0.05; fig 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (SE) TNFα protein levels in BAL fluid cell culture supernatants of patients after 18 hours without stimulation (18 h) and following stimulation with rhIL-10 (+rhIL-10) or rhIL-13 (+ rhIL-13) as well as stimulation with lipopolysaccharide (LPS) with and without rhIL-13. *p<0.05 v unstimulated cells; **p<0.05 v LPS alone.
DISCUSSION
Inflammation in sarcoidosis is mainly driven by Th1 cytokines such as IL-1, IL-2, IL-6, IFN-γ, or TNFα which have proinflammatory effects.2–5 Since patients often have only mild symptoms, counteracting mechanisms such as anti-inflammatory cytokines have been suggested. However, information on these cytokines in sarcoidosis is sparse. Soluble TNF-R, TGFβ1, and IL-10 have been investigated as possible counteracting mediators. Increased levels of TNF-Rs have been seen in the plasma and BAL fluid of patients with sarcoidosis.11 Armstrong et al12 showed that TNFα bioactivity in the lungs of subjects with sarcoidosis may be inhibited by increased levels of soluble TNF-R. This inhibition may be greater in stage I disease than in stage II/III, indicating a homeostatic mechanism which protects the lung from excessive TNFα production. Increased concentrations of TGFβ1 have been reported in cell culture supernatants of BAL fluid cells.13 Enhanced tissue localisation of TGFβ1 and related extracellular matrix proteins with granulomas of sarcoidosis has also been seen, which suggests that TGFβ1 may modulate the fibrotic repair process accompanying granuloma healing.23 TGFβ1 expression may be involved in the regression of granulomas of sarcoidosis by initiating fibrosis and atrophy of epithelioid cells.24 However, increased TGFβ1 levels have also been observed in the BAL fluid of sarcoidosis patients with altered lung function, supporting the hypothesis that TGFβ1 overproduction is associated with lung function impairment in these patients.14 IL-10 secretion has been reported to be increased in alveolar macrophages15 but not in BAL fluid cells of patients with sarcoidosis.13
IL-13 is suitable for investigating patients with sarcoidosis for several reasons: (1) it is mainly produced by CD4+ T cells which are also increased in the BAL fluid of sarcoidosis patients; (2) patients with sarcoidosis suffer from bronchial hyperreactivity more often than the general population (23% versus 11–14%)25; and (3) IL-13 has been shown to inhibit TNFα production in blood monocytes.21
In this study IL-13 mRNA expression was significantly increased in BAL fluid cells and PBM of patients with sarcoidosis compared with controls. This effect might be mediated in part by IL-13 since this mediator has been shown to be involved in the pathogenesis of bronchial hyperresponsiveness20 which occurs more frequently in patients with sarcoidosis than in the general population.25 The inflammatory reaction found in the asthmatic lung is mediated by Th2 cytokines such as IL-13 and IL-4. No significant difference in IL-4 expression in BAL fluid cells and PBM was found between the patients and controls, which suggests that IL-4 does not contribute to the pathophysiological reactions in sarcoidosis. This finding supports the notion that IL-13 might be a central mediator leading to increased bronchial reactivity in a subgroup of sarcoidosis patients. Moreover, increased levels of IgE serum proteins are frequently found in these patients. IL-4 does not seem to cause increased IgE concentrations because IL-4 expression was not increased in PBM of patients. We also found increased IgE concentrations in 10 patients (data not shown) but there was no significant correlation between IgE serum levels and IL-13 (or IL-4) expression in PBM. However, this finding does not rule out the possibility that increased expression of IL-13 in PBM might cause increased IgE serum protein levels, thus contributing indirectly to bronchial hyperreactivity in sarcoidosis.
As expected, the percentage of lymphocytes was higher in the patient group than in the control group. This might contribute to increased IL-13 expression in the patient group, but Prasse et al26 recently reported that IL-4 and IL-13 expression in BAL T cells measured by flow cytometry was not increased in patients with sarcoidosis. Moreover, in our study IL-13 expression in PBM was increased in patients, although the percentage of lymphocytes was lower than in the control group (20% v 57%). We measured mRNA expression rather than protein levels, but lymphocytes do not seem to be the major source of IL-13 expression in sarcoidosis. Immunocytochemistry of BAL cells from eight patients with sarcoidosis showed that about 30% of alveolar macrophages stained positively for IL-13 whereas only a few lymphocytes were weakly positive. Since alveolar macrophages have been shown to express IL-13 in pulmonary fibrosis,22 they might also express IL-13 in other interstitial lung diseases. Our data suggest that alveolar macrophages might be an important source of IL-13 in sarcoidosis.
IL-13 protein levels were slightly increased in BAL cell culture supernatants in some patients, but the difference between the patients and controls was not statistically significant. This might be because of the small sample size and the low levels of IL-13. However, half of the patients investigated had increased IL-13 concentrations, indicating that IL-13 is released by BAL fluid cells in sarcoidosis.
As expected, TNFα mRNA expression was significantly increased in BAL fluid cells of patients compared with control subjects, while no significant difference was seen in PBM. This finding is in agreement with previous data by Steffen et al.5 TNFα might be responsible for inflammatory clinical reactions but the course of the disease in sarcoidosis is often mild. It is possible that other anti-inflammatory mediators might counteract TNFα.9 IL-10 has been shown to suppress TNFα,27,28 but in our study IL-10 expression could not be detected in most of the patients. Nevertheless, in a study by Minshall et al29 an increase in IL-10 mRNA positive BAL cells was seen in patients with sarcoidosis who had recently developed increasing cough or dyspnoea, systemic symptoms and/or increasing opacities on the chest radiograph indicating an active state of disease. Zissel et al13 reported no spontaneous IL-10 release from BAL fluid cells of patients with sarcoidosis. In contrast, Bingisser et al15 recently described increased local secretion of IL-10 from alveolar macrophages in sarcoidosis. The different results may derive from the cell populations studied (BAL cells or alveolar macrophages). Further studies are needed to define the role of IL-10 in the pathophysiology of sarcoidosis.
To investigate the effect of IL-13 on TNFα production, BAL cell cultures from patients were incubated with rhIL-13, with and without LPS stimulation. The mean TNFα concentration was decreased in cell culture supernatants in the presence of IL-13; this effect was even greater in LPS stimulated BAL cell cultures. Cosentino et al21 showed that IL-13 could decrease TNFα secretion in blood. Our data suggest that IL-13 might inhibit TNFα production in BAL cells in sarcoidosis. Incubation with rhIL-10 also decreased TNFα protein significantly, demonstrating the ability of IL-10 to reduce proinflammatory cytokine expression.
Comparison of patients with stage I disease and those with stage II/III disease revealed higher expression of IL-13 mRNA in BAL fluid cells of patients with stage I disease. Stage I is defined by the presence of bilateral hilar lymphadenopathy (BHL) with no interstitial changes; in stage II BHL is associated with nodular shadowing; and in stage III there is more generalised shadowing without BHL. Stage I is usually a self-limiting disease whereas stages II and III may be associated with a more progressive course. These findings suggest that IL-13 may be involved in early inflammation in sarcoidosis and may inhibit proinflammatory reactions, thus supporting limitation of disease. IL-13 may not play an important role in more advanced disease with structural changes in the lungs. It has recently been shown that IL-13 stimulates TGFβ1 production in transgenic mice, indicating that the fibrogenic effects of IL-13 are mediated to a great extent by a TGFβ1 pathway.30 It is tempting to speculate that IL-13 might also activate TGFβ1 in sarcoidosis, leading to regression of sarcoid granulomas.
IL-13 has been shown to be a promising modality in the treatment of uveitis,31 and intratracheal instillation of IL-13 was found to have anti-inflammatory activity in the airways of TNFα- or antigen-challenged guinea pigs.32 By provoking airway hyperresponsiveness, IL-13 may not be a suitable therapeutic agent for treating lung disease in sarcoidosis, but it may help to restore the disturbed Th1/Th2 balance in these patients. Another possible option might be selectively to induce anti-inflammatory activity via the IL-13 receptor.
In conclusion, our data suggest that IL-13 might play a role as an anti-inflammatory mediator in pulmonary inflammation in sarcoidosis by acting on TNFα. Further studies are needed to determine whether IL-13 influences other mediators of inflammation in sarcoidosis and whether it is associated with the fibrotic process because overexpression of IL-13 in an animal model resulted in subepithelial fibrosis.33 Future studies should also investigate whether IL-13 might contribute to airway hyperresponsiveness in sarcoidosis. This would clearly determine whether IL-13 can be considered a possible treatment option in this disease.
Acknowledgments
The authors thank Dr Rame A Taha for carefully reading this manuscript and for instructive discussion.
REFERENCES
Footnotes
-
Hans-Peter Hauber is supported by a scholarship from the German Cystic Fibrosis Foundation.
Linked Articles
- Airwaves