Article Text

Abstract
Background: Surfactant synthesis and secretion has been shown to be impaired in type II cells from diseased lungs. The mechanism of surfactant lipid recycling, which is an important physiological process in surfactant treatment, was studied in type II cells isolated from injured lungs.
Methods: Different stages of lung injury were induced by exposing rats to 10 ppm nitrogen dioxide (NO2) for 3, 20, and 28 days. Type II cells were isolated from these lungs and recycling of 3H-DPPC labelled surfactant-like liposomes was studied in vitro.
Results: Uptake of liposomes (150 μg/ml) for 20 minutes in the absence and presence of surfactant protein-A (SP-A, 5 μg/ml) was higher in cells from NO2 injured lungs (63–78%) than in control cells. There was no difference in liposome uptake between the groups with NO2 exposure of different duration. After liposome uptake, most of the internalised label remained in the phosphatidylcholine (PC) fraction and increased with duration of exposure to NO2. After 20 minutes internalisation, cells were allowed to resecrete lipids for a further 20 minute period. In cells from controls and from all stages of lung injury, liposomes that had been internalised in the presence of SP-A were resecreted to a greater extent than those internalised without SP-A. However, cells from lungs exposed to NO2 resecreted less lipid than cells from control lungs. Again, there was no difference in resecretion between the groups with NO2 exposure of different duration.
Conclusion: Type II cells from injured lungs internalise more surfactant-like liposomes than cells from controls, suggesting a putative therapeutic significance to cope with limited alveolar surfactant pools in lung injury.
- surfactant system
- nitrogen dioxide
- type II pneumocytes
- bronchoalveolar lavage
- surfactant-like liposomes
Statistics from Altmetric.com
Pulmonary surfactant is a surface tension lowering lipid-protein complex lining the surface of the alveoli and preventing the lung from atelectasis during end expiration. It is synthesised and secreted into the alveolar lumen by alveolar type II pneumocytes.1 Because surfactant secretion is a permanent process, there are clearance mechanisms that prevent the alveoli from being filled with this material, thus guaranteeing a relatively stable surfactant concentration. In normal lungs, possible mechanisms for surfactant clearance from the alveoli include its removal via airways, blood, and lymph, its degradation in the alveoli, or its reuptake into cells of the airways, into macrophages, and into cells of the respiratory epithelium.2 In vivo experiments in normal lung tissue have shown that surfactant material taken up from the alveolar lumen is directed to type II cells.3,4 In primary type II cell culture this clearance pathway was also observed for phosphatidylcholine (PC), the major phospholipid moiety.5
A number of lung diseases involve alteration of the pulmonary surfactant. The most important is the infant respiratory distress syndrome (IRDS) in which the alveoli lack surfactant material, thus bearing the risk of life threatening situations.6 In other lung disorders the composition and metabolism of the surfactant are impaired—for example, the acute respiratory distress syndrome of the adult (ARDS) and oxidant induced lung injury.7,8 For some years the endotracheal instillation of exogenous surfactant has been an accepted treatment to overcome the lack of an appropriate amount of this material in patients with IRDS.9 Treatment of other lung diseases in which surfactant is involved are under discussion, but a detailed evaluation of the impaired metabolism and recycling of surfactant is needed.
In diseased lungs the only information available to date is on the synthesis of pulmonary surfactant in type II pneumocytes that have been isolated from oxidant injured lungs. Surfactant synthesis was found to be increased in cells from nitrogen dioxide (NO2) and hyperoxia injured lungs,10–12 while secretion of surfactant from NO2 injured lung cells was reduced.13 However, there is no information available on the uptake and resecretion of surfactant in type II pneumocytes from diseased lungs. Such information is an essential prerequisite for understanding the pathophysiology and development of treatment strategies to increase the intracellular surfactant pool in conditions in which extracellular surfactant is altered.
The present study was designed to obtain information on uptake of surfactant-like liposomes and lipid resecretion from type II pneumocytes of differently injured lungs. To induce different stages of lung injury experimentally, rats were exposed to continuous inhalation of 10 ppm NO2 for 3, 20, and 28 days in order to induce bronchitis and emphysema. The type II pneumocytes isolated from the different stages of lung injury were analysed for vitality, their capacity to internalise surfactant-like liposomes, and to resecrete lipids.
METHODS
Animals, NO2 exposure and experimental groups
All experiments were carried out on specific pathogen free adult male Sprague Dawley rats (body weight 170–180 g) obtained from Charles River Wiga (Sulzfeld, Germany). Every experiment was done with 8–12 animals. Each experimental group consisted of four animals that were kept in one exposure chamber with free access to food and water.
Nitrogen dioxide atmospheres were generated by mixing compressed air with NO2 from a tank (Messer-Griesheim, Duisburg, Germany). The final concentration for all exposure experiments was 10 ppm. Normal air breathing animals were used as controls because we have found earlier that there is no difference in these and those exposed to compressed air.
Chemicals
All reagents were purchased from Sigma (Deisenhofen, Germany) unless stated otherwise. The Dulbecco’s modified Eagles’s medium (DMEM) for culture of type II cells was supplied by Life Technology (Karlsruhe, Germany) and radiolabelled lipids were purchased from Amersham (Braunschweig, Germany).
Isolation of type II pneumocytes
Rats were anaesthetised with pentobarbital sodium (50 mg/kg) mixed with 100 IU heparin sodium and injected intraperitoneally. When the animals were in deep anaesthesia the trachea was cannulated and, after thoracotomy, the vena cava was cut and the lungs perfused free of blood via the pulmonary artery and removed from the body. The lungs were washed extracorporally with five volumes of 8 ml normal warm (37°C) saline; after instillation of each volume the fluid was allowed to run out passively. To obtain alveolar cells and debris free lavage the fluid was centrifuged at 300g and 4°C for 10 minutes and the resulting fractions were kept for further determinations.
Isolation of type II cells was done following the procedure described by Dobbs and coworkers.14 In brief, after bronchoalveolar lavage the lungs were washed with the solutions described by Dobbs before elastase solution was instilled. Digestion with elastase was allowed to take place at 37°C for 20 minutes before the large airways were removed; in the presence of DNase I (250 μg/ml) the lungs were minced with scissors and the elastase reaction was then stopped by addition of 5 ml fetal bovine serum per lung (Gibco-BRL, Eggenstein, Germany). The final cell suspension was filtered several times through gauze nylon and washed by gentle centrifugation. The cell pellet was resuspended in DMEM and transferred to rat immunoglobulin G coated bacteriological Petri dishes to a density of 30 × 106 cells. After incubation for 1 hour at 37°C in a 10% CO2-air incubator, the macrophages were adherent to the plastic dishes. The unattached type II pneumocytes were removed, centrifuged, and used for further experiments.
Purity and viability
After cell isolation, type II cell viability was determined by trypan blue dye exclusion. The purity of the cell preparation was evaluated with the Papanicolaou stain. Samples of every exposure condition were evaluated per microscope field in triplicate and an average value was determined.
Isolation of lamellar bodies
The isolation of lamellar bodies was performed as outlined elsewhere.28 In brief, type II cells were homogenised in 0.32 M sucrose in 10 mM Tris/NaCl buffer and layered over a discontinuous gradient of 0.45 M and 0.55 M sucrose in Tris/NaCl buffer. After centrifugation at 90 000g for 3 hours, the band at the interface between 0.45 M and 0.55 M sucrose was collected. The lamellar body was obtained after dilution of the interface to 0.2 M sucrose followed by centrifugation at 20 000g for 15 minutes.
Liposome preparation
For studies of surfactant-like liposome uptake, unilamellar liposomes were prepared as described by Wissel et al,15 mixing 5.5 mg 1,2-dipalmitoyl-l-3-phosphatidyl-N-(methyl-H) choline (DPPC), 1 mg PG, 2.5 mg egg yolk PC, and 1 mg cholesterol in chloroform. This mixture was supplemented with 3H-DPPC (specific activity 87 μCi/mg) to a final radioactivity of 1.46 nCi/μg lipid. After evaporation of the chloroform, 1 ml 0.2 M phosphate buffered saline (PBS, pH 7.4) was added and the mixture was incubated at 50°C for 15 minutes. The lipid mixture was pulse sonicated (Brenson B12 Sonofier, Heinemann Schwäbisch-Gemünd, Germany) at 40% maximal output for 20 minutes. For resecretion experiments liposomes were prepared as described above with the following composition: 5.4 mg DPPC, 1 mg PG, 2.5 mg egg yolk PC, 1 mg cholesterol, and 0.1 mg cholesterol oleate in chloroform.15 This mixture was supplemented with 3H-DPPC (specific activity 87 μCi/mg) to a final radioactivity of 1.46 nCi/μg lipid and with 14C-cholesterol oleate (specific activity 115 mCi/mg) to a final radioactivity of 1 nCi/μg lipid. The mixture was stored at 4°C and used within 1 week. Before use the liposomes were centrifuged at 1000g to remove larger aggregates.
Liposome uptake into isolated type II cells
Liposome uptake was studied using freshly isolated type II cells in floating culture. Cell culture tubes with 2.5 × 106 cells in 1 ml DMEM containing 0.1% (w/v) lipid free bovine serum albumin (BSA) were incubated with 9 μl of the described liposome suspension (see above) in the absence or presence surfactant protein-A (SP-A, 5 μg/ml) over 60 minutes at 37°C. Samples were taken every 10 minutes to determine the kinetics of liposome uptake. Cells were sedimented by centrifugation (160g, 4°C, 10 minutes) and resuspended in DMEM containing 5% fat free BSA and 10 mM ethylene glycol-bis (β-aminoethyl ether)-N,N,N1N1-tetraacetic acid (EGTA). Sedimentation and resuspension were repeated twice in DMEM containing 0.1% BSA and 10 mM EGTA and a final washing procedure with only DMEM. Lipid material still associated with the type II cells after this rigorous washing procedure is termed “internalised lipids” or lipid uptake.
Thin layer chromatography
To facilitate recovery and determination of internalised liposomes, carrier lipids (organic lipid extract from pig lung homogenate, 600 μg, dissolved in 100 μl chloroform/methanol 2:1) were added to the samples. After lipid extraction,16 phospholipids were separated by two dimensional thin layer chromatography (TLC) on silica H60 TLC plates (Merck, Mannheim, Germany) as described by Wissel et al.17 The phospholipid spots were visualised by brief exposure to iodine vapour, scraped into 6 ml Optifluor (Packard, Groningen, Netherlands) scintillation fluid, and counted for radioactivity.
For experiments comparing liposome uptake into cells from lungs exposed to NO2 for different durations, an uptake period of 20 minutes was used. Because the amount of phospholipid from the single experiments was low, phospholipids were only analysed for the PC and “other lipids” moieties.
Surfactant resecretion from isolated type II pneumocytes
After liposome uptake, the radioactivity in the washed type II cells represents the net liposome uptake—that is, 100% radioactivity at time point zero for the resecretion process.
As in the studies of liposome uptake, an optimal period of resecretion had to be determined with kinetic studies. After the liposome internalisation process, cells from control animals and from those exposed to NO2 were resuspended in 1 ml DMEM containing 0.1% BSA and incubated at 37°C for 60 minutes. Samples were taken every 10 minutes to determine the resecreted lipid in the media and the remaining label in the cells. The resecretion process was terminated by addition of 1 ml ice cold DMEM. Further cell washing proceeded as described previously. All supernatants were pooled and kept for determination of the resecreted radioactive material. Phospholipid extraction was performed as described for liposome uptake.
Isolation of surfactant associated protein (SP-A)
Surfactant associated protein A (SP-A) was isolated from lung lavages from normal control rats using the method of Hawgood and coworkers.18 Protein purity was analysed in SDS PAA gels and bioactivity as the capacity to inhibit phosphatidylcholine secretion from isolated type II cells.
Other methods
Protein was measured with the Bio-Rad reagent (Munich, Germany)19 and phospholipid was calculated according to standard procedures.20 Lactate dehydrogenase (LDH) activity was measured in cells and culture media by conversion of β-nicotinamide adenine dinucleotide (β-NAD).21
Statistical analysis
Assays were performed in triplicate for each sample from each exposure condition and a mean value was determined. The mean values for each sample were then used for analysis. The results are expressed as mean (SE) values. For paired analyses between two groups the Student’s t test was used. Statistical significance between more than two groups was tested using the ANOVA procedure and subsequent Scheffé test. p values of ⩽0.05 were considered significant.
RESULTS
Type II cell preparations, cell yield, purity and viability
Evaluation of in vitro surfactant-like liposome uptake and lipid resecretion from type II pneumocytes from lungs exposed to NO2 for different durations was only performed on freshly isolated cells. As previously reported,22 the yield of type II pneumocytes from NO2 exposed lungs was significantly higher than that from control lungs (data not shown). Cell purity determined by Papanicolaou stain was comparable for all cell preparations and ranged from 91% to 93% (n=44 cell isolations). Cell viability as evaluated by trypan blue dye exclusion was also comparable in all preparations (range 95–96%, n=44 isolations).
Kinetics of liposome association
To determine an incubation period for a comparison of the association of surfactant-like liposomes with type II cells from all experimental groups (0, 3, 20, and 28 days of NO2 exposure), the uptake kinetics were evaluated at 37°C over a 60 minute time period. During this period type II pneumocytes (2.5 × 106 cells/ml) were incubated in DMEM with 3H-DPPC containing liposomes (150 μg/ml) in the absence and presence of 5 μg/ml SP-A. Cell aliquots were taken from the samples every 10 minutes, subjected to the rigorous washing procedure to remove lipid adherent to the cells, and assayed for associated radioactivity.
In cells isolated from the lungs of control animals in the absence of SP-A, the amount of 3H-labelled liposomes increased faster at early time points (10, 20, and 30 minutes) than at 40 and 60 minutes (fig 1). Liposome association with cells from lungs exposed to NO2 for 3 days followed the same kinetics, but the amount of cell associated lipid for all time points measured was significantly increased compared with control cells. Liposome uptake into cells exposed to NO2 for 20 and 28 days had the same kinetics as those for cells exposed for 3 days (because the graphs overlapped with the 3 day NO2 exposure data curve they were not included in the figure). When liposome uptake was performed in the presence of SP-A (5 μg/ml), the cell associated label followed the same kinetics but was about three times higher than in the experiments without SP-A. At 20 minutes the cell associated liposome label was about half maximal and this was therefore regarded suitable for further comparative studies.
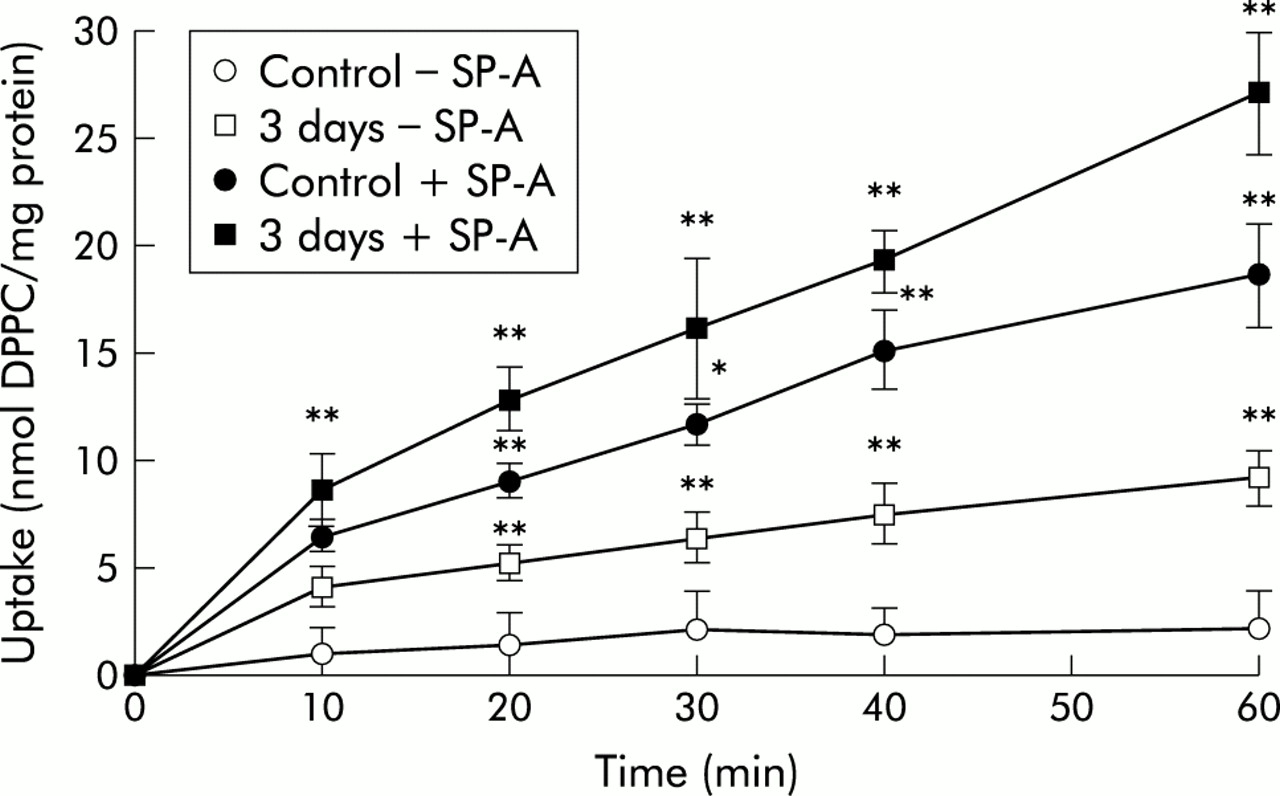
Time course of uptake of 3H-DPPC labelled liposomes by freshly isolated type II cells from control animals and those exposed to 10 ppm NO2 for 3 days. Type II cells were isolated from lungs of controls and from lungs with different stages of lung injury induced by exposure to atmosphere containing 10 ppm NO2 for 3, 20, and 28 days. Because the graphs of uptake kinetics for the cells exposed to NO2 for 20 and 28 days overlapped with those of the 3 day exposure, they were not included in the figure. Values are mean (SE), n=5; *p⩽0.05, **p⩽0.01 difference between time points of the 3 day exposure to NO2 and corresponding time points of control lungs in the absence and presence of SP-A.
Is liposome association “adherence to cell membranes” or “cellular uptake”?
To determine whether labelled liposomes were internalised into cells or just adhered to the cell membranes, the incubation assay was also performed at low temperature (4°C) to block energy dependent active uptake processes. After incubation for 20 minutes a considerable amount of strongly adherent liposome material was attached to the cells that could not be removed by normal DMEM washings. Using the rigorous washing procedure (three washing cycles in DMEM supplemented with 5% fat free BSA and 10 mM EGTA solution followed by two washing cycles in DMEM with 0.1% BSA), the adherent liposome material was reduced to 40% of the attached material. Further washings did not remove any additional liposomes. The labelled liposomes that were resistant to the rigorous BSA-EGTA washing procedure are therefore considered as cellular liposome uptake in this study.
After the washing procedure a considerable amount of label from the liposomes was associated with the cells at 4°C, suggesting that lipid exchange makes a significant contribution to the apparent lipid uptake or non-calcium dependent adherence. Because of this possibility, liposomes were labelled with cholesterol oleate, a lipid that does not easily exchange, as suggested elsewhere.15 For all experimental groups liposome uptake in the absence of SP-A did not differ whether it was performed at 4°C or 37°C (table 1). When SP-A was included in the uptake experiments at 4°C, comparable values were obtained to those in the experiments without SP-A at 4°C and 37°C. A clear increase in liposome uptake was observed when SP-A mediated uptake was performed at 37°C, resulting in an increase of label in the cells from control and from NO2 exposed lungs of 2–3 fold.
Temperature dependency of liposome uptake by type II pneumocytes isolated from lungs exposed to 10 ppm NO2 under different conditions
Lipid uptake into cells from diseased lungs
As shown in fig 2, after incubation for 20 minutes at 37°C, uptake of 3H-DPPC labelled liposomes into type II cells from the different periods of NO2 exposure (3, 20, and 28 days) was increased compared with uptake by control cells (p<0.05 for 3, 20 and 28 days v no exposure). The increase was similar for all periods of NO2 exposure and was 68–81% higher than that of control cells; there was no difference between the different NO2 exposure groups. When SP-A (5 μg/2.5 × 106 cells) was included in the incubation medium, uptake of DPPC labelled liposomes into cells from control and NO2 exposed lungs was more than twice that without SP-A. Internalisation into cells from NO2 exposed lungs was 63–78% more than that of control cells, with no difference between the different NO2 exposure groups.

Uptake of 3H-DPPC labelled liposomes into isolated type II pneumocytes from lungs of control animals and from those exposed to 10 ppm NO2 for 3, 20, and 28 days. The white bars represent liposome uptake in the absence of SP-A, the black bars represent liposome uptake in the presence of SP-A. Values are mean (SE), n=3–5; *p⩽0.05, **p⩽0.01 difference between time points of the 3 day exposure to NO2 and corresponding time points of controls in the absence and presence of SP-A.
As already described, at low temperatures an increased 3H-DPPC liposome label was associated with the cells, suggesting the possibility of lipid exchange to the apparent uptake. To determine whether the observed lipid uptake was due, at least in part, to lipid exchange, uptake experiments were performed with 3H-cholesterol ester labelled liposomes, a lipid marker that does not exchange easily.
As shown in fig 3, the results from these experiments were comparable to those with 3H-DPPC labelled liposomes. For all NO2 exposed groups in the absence of SP-A, a 75% increase in 14C-cholesterol ester labelled liposome uptake was found compared with cells from control lungs. In the presence of SP-A, the uptake of 3H-cholesterol ester labelled liposomes was about 250% higher than in the absence of SP-A. Cells from NO2 exposed lungs incorporated about 75% more cholesterol ester than cells from the unexposed lungs.

Uptake of 14C-cholesterol oleate labelled liposomes into isolated type II pneumocytes from lungs of control animals and from those exposed to 10 ppm NO2 for 3, 20, and 28 days. The white bars represent liposome uptake in the absence of SP-A, the black bars represent liposome uptake in the presence of SP-A. Values are mean (SE), n=3–5; *p⩽0.05, **p⩽0.01 difference between time points of the 3 day exposure to NO2 and corresponding time points of controls in the absence and presence of SP-A.
To confirm cell viability after liposome incubation, cells were assayed by trypan blue dye exclusion and by measurement of lactate dehydrogenase (LDH) activity in the incubation media. Cell viability was comparable in all experimental groups with a range of 93–97% (n=38 experiments). In all incubation experiments after 20 minutes of liposome uptake, LDH activity in the media was less than 2% of the total cellular activity (n=38 experiments).
Liposome label in lamellar bodies after NO2 exposure
After liposome uptake into type II cells, the cell associated label was higher in cells that had been incubated with liposomes in the presence of SP-A than in those incubated in the absence of SP-A. To determine the extent to which radioactivity was contained in the lamellar bodies, these subcellullar elements were isolated from the 0.45 M sucrose gradient centrifugation fraction and determined for radioactivity.
It was found that liposome uptake resulted in higher lipid label in the lamellar bodies in the presence of SP-A than in the absence of SP-A (table 2). This SP-A mediated increased uptake was threefold for type II cells from controls and twofold for cells from the different NO2 exposure groups. Even in the absence of SP-A, cells from NO2 exposed animals incorporated more label into the lamellar bodies than cells from control animals. There was no difference in the radioactivity of the lamellar bodies in cells from control animals and those in the different NO2 exposure groups.
Effect of exposure to 10 ppm NO2 on incorporation of radioactivity into lamellar bodies of type II pneumocytes
Kinetics of lipid resecretion
To ascertain whether type II cells from the different NO2 exposure groups resecrete lipids from the internalised liposomes, an optimal resecretion period had to be determined to allow comparison between all experimental groups. Freshly isolated type II pneumocytes were incubated in the presence and absence of SP-A (5 μg/ml) for 20 minutes at 37°C with 3H-DPPC labelled liposomes as described earlier. For all experimental groups the amount of internalised 3H-DPPC labelled liposomes was set at 100%. After liposome uptake, cells were washed with BSA-EGTA supplemented DMEM to remove adherent lipid and incubated in lipid free medium for 60 minutes. During this period type II cells were allowed to resecrete the incorporated lipid material. Resecretion was calculated as the percentage of resecreted label in the medium and the label in the medium plus that in the cells.
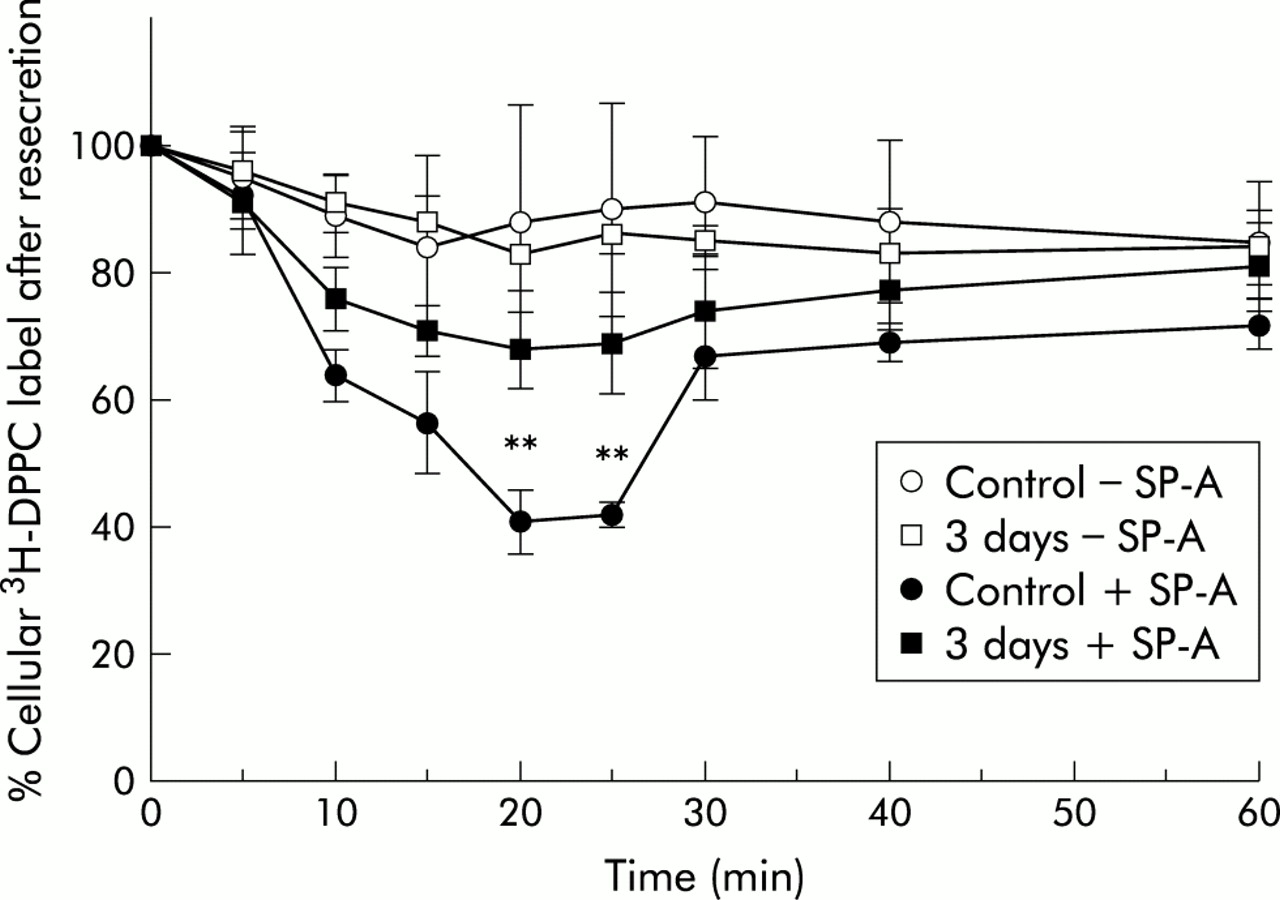
Resecretion kinetics showed that, in cells from control lungs and those exposed to NO2 for 3 days, lipids internalised in the absence of SP-A were resecreted over 60 minutes only to a small extent (10–15% of internalised material; fig 4). Cells from lungs exposed to NO2 for 20 and 28 days followed the same kinetics (graphs were not included in the figure because of overlapping with the 3 day NO2 exposure curve). There was no obvious difference in lipid resecretion for cells from control lungs and those from NO2 exposed lungs. In contrast, lipids that had been internalised in the presence of SP-A were resecreted to a greater extent. The intracellular amount decreased significantly with time until 20 minutes resecretion. At this time point resecretion from cells of control lungs had reached a maximum (59% of the intracellular label). After that period the intracellular label increased again, reaching 74% of the original amount of internalised label. The cells from the NO2 exposed lungs followed the same kinetics (graphs for 20 and 28 day NO2 exposure not included in the figure because of overlapping). However, type II cells from NO2 exposed lungs resecreted less lipid material than cells from control lungs. Because maximum resecretion occurred in all experimental groups at 20 minutes, this time point was regarded as suitable for comparison of lipid resecretion between all experimental groups.

Time course of resecretion of 3H-DPPC labelled liposomes from isolated type II cells from control animals and animals exposed for 3 days to 10 ppm NO2. The data represent the intracellular label as a percentage of the internalised material. Values are mean (SE), n=3–5; *p⩽0.05, **p⩽0.01 difference between time points of the 3 day exposure to NO2 and corresponding time points of control in the absence and presence of SP-A.
Lipid resecretion from type II cells of diseased lungs
As shown in fig 5, at 37°C resecretion of DPPC that was internalised in the absence of SP-A was significantly reduced from lungs exposed to NO2 for 3, 20, and 28 days compared with controls (p<0.01 for 3, 20, and 28 days v controls). No differences were observed between the different NO2 exposure groups. Up to 58% of the amount of liposomes that were internalised by control cells were resecreted compared with 10% in cells from the NO2 exposed lungs. From the 3H-DPPC labelled lipids that were internalised in the presence of SP-A, a larger quantity was resecreted from the lipids taken up without the protein. Furthermore, for resecretion of the SP-A mediated internalised liposomes it was found that lipid resecretion was lower in cells from NO2 exposed lungs than from controls, but the rate of resecretion did not differ between exposure groups.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Resecretion of internalised lipids from isolated type II cells from control animals and animals exposed to 10 ppm NO2 for 3, 20, and 28 days. The white bars represent remaining intracellular lipids from liposomes that have been internalised in the absence of SP-A, the black bars represent remaining intracellular lipids from liposomes that have been internalised in the presence of SP-A. The data represent the remaining intracellular label after lipid resecretion. Values are mean (SE), n=4; *p⩽0.05, **p⩽ 0.01 difference between time points of the 3 day exposure to NO2 and corresponding time points of control in the absence and presence of SP-A.
Cell viability as evaluated by trypan blue dye exclusion exhibited a viability of 95–98% for all experimental groups. In addition, LDH activity in the cell culture media of the resecretion experiments was less than 3% of the total cellular activity.
Distribution of label from 3H-DPPC incorporated liposomes among lipid classes and fate of resecreted lipids
To determine whether 3H-DPPC labelled liposomes were metabolised by type II cells during the uptake period, the distribution of the intracellular label was determined. After phospholipid extraction and two dimensional TLC, phosphorus was measured and radioactivity associated with the PC moiety and other phospholipid moieties determined. For all NO2 exposed groups, incorporation of 3H-DPPC into the PC moiety was significantly increased compared with uptake into the PC fraction of control cells (table 3).
Fate of internalised and resecreted 3H-DPPC containing liposomes
To determine whether lipids from 3H-DPPC labelled liposomes that were internalised in the absence and presence of SP-A were modified by type II cells from various stages of NO2 induced lung injury, the distribution of the label in the PC moiety and in other phospholipid moieties was determined. Freshly isolated type II cells were allowed to internalise and resecrete lipids as described above. After resecretion, phospholipids were extracted from the cells and the media and separated by two dimensional TLC. Phosphorus was measured and radioactivity associated with the PC moiety and other phospholipid moieties determined.
In cells from control and from NO2 exposed lungs in the absence of SP-A, most of the liposomal DPPC that was internalised was found in the PC moiety. There was a linear trend for the percentage of label to increase with the duration of NO2 exposure. This increase in PC label was significant for cells exposed to NO2 for 20 and 28 days. Label from the DPPC liposomes taken up by the SP-A mediated pathway was also incorporated into the PC moiety, but to a greater extent than in assays without SP-a. In these experiments, type II cells from NO2 exposed lungs incorporated a greater amount of label than cells from control lungs.
After lipid resecretion, most of the remaining intracellular label stayed with the PC but most of the extracellular label was found in the non-PC lipid moiety. Resecretion of lipid from SP-A mediated liposome uptake resulted in an unmodified distribution of label from internalised 3H-DPPC. More than 90% of the label remaining within the cells, as well as the extracellular label, was still associated with the PC moiety.
DISCUSSION
There is evidence from a number of studies that type II pneumocytes in normal lungs represent the major compartment for surfactant clearance from the alveolar space.1 However, there is no information on this process in diseased lungs. This mechanism is of interest for the therapeutic application of surfactant in injured lungs where it should be internalised by type II cells. The aim of the study was therefore to elucidate the extent to which type II cells from different stages of lung injury are able to internalise and resecrete surfactant lipids.
To experimentally induce lung injury, rats were exposed over different time periods to NO2. This procedure was found to generate different stages of lung injury showing histological correlates with acute and chronic bronchitis (after 3 and 20 days inhalation of 10 ppm NO2) and emphysema (after 28 days),23 comparable to the situation in humans. From all stages of lung injury sufficient amounts of type II cells were obtained with a purity of 91–93% as evaluated by the Papanicolaou stain. From LDH measurements it was found that the permeability of the cells from all experimental groups was not impaired, indicating that only fully vital cells were used. Throughout the experiments cell viability was comparable for all experimental groups (range 94–95%). We therefore feel confident that the data obtained from our assays were from intact/viable cells rather than from degenerating cells.
Uptake of surfactant-like liposomes by type II pneumocytes in the presence of SP-A has been studied previously. However, with confocal laser scanning microscopy techniques it was recently shown17 that, after incubation with liposomes, measurable amounts can be localised within the cells but only a small amount of material is attached to the cell membrane. To ensure that the cells also internalised the lipid material, we used the same uptake method as that described by Wissel et al.17 Apart from use of an appropriate uptake assay, it was necessary to determine whether lipid exchange and energy dependent internalisation in the absence and presence of SP-A contribute to the cell associated label. Type II cells from control and NO2 exposed lungs were incubated with cholesterol ester, a lipid marker that does not exchange easily, and it was found that, in the absence of SP-A, a certain amount of lipid results either from lipid exchange or from adherence to the cell membrane. This process does not require energy, as shown by the high association of cholesterol ester at 4°C. However, in the presence of SP-A, type II cells actively internalise a considerably larger amount of lipid. This was energy dependent because low temperature (4°C) reduced the SP-A mediated lipid uptake to the level found for unstimulated uptake (table 1). This effect was observed for cells from both control and NO2 exposed lungs. It is clear that lipid uptake in these cells is composed of an active and a passive part. From this and results reported by other groups,17,24 it appears that the energy dependent SP-A mediated uptake contributes most of the lipid uptake. The different stages of NO2 induced lung injury did not influence the relation between active and passive lipid uptake. In addition, accumulation of radioactivity in the lamellar bodies indicated lipid internalisation rather than simply adherence to the cell membrane.
As found in the kinetic experiments of liposome uptake (fig 1), there is a faster uptake into the cells from NO2 exposed lungs than in those from controls. The finding that there is no difference in the uptake kinetics between the cells from the different NO2 exposure groups suggests that the maximal effect was achieved by 3 days and could not be increased further by 20 and 28 days of exposure. The basis for this increase, however, is not known, but it is possible that the increased internalised liposome material is used for phospholipid synthesis. For type II cells from acutely NO2 injured lungs, an increased uptake and incorporation of choline into phospholipid was observed based on an increase in the specific activity of the choline kinase that showed increased phospholipid synthesis.11
Although in vitro experiments have suggested that a number of functions in lipid metabolism have been directed to SP-A, including the stimulation of lipid uptake,25 in vivo the situation is different because a number of SP-A attributed functions in vitro could not be found in vivo in SP-A gene targeted mice.26 In later life, however, the size of the surfactant pool of wildtype and knock out mice is no longer different.27
As shown in fig 2, type II cells from all groups of NO2 exposed lung show increased liposome uptake compared with control cells. In these figures we preferred the calculation of lipid per cells rather than per protein because it is known that cell protein varies in cells. The fact that this increase was similar in the cells of all exposed lungs suggests that NO2 exposure may have disrupted cell membranes to allow liposome to enter the cells easily. However, LDH and trypan dye exclusion measurements before and after the washing procedures and incubation periods showed no cell degeneration. We therefore conclude that a mechanism is activated by the injury which leads to accumulation of lipid within the cells of NO2 exposed cells, but the nature of such a postulated mechanism is not known. An increased uptake into the cells from the NO2 exposed animals was also observed with the cholesterol ester (fig 3). Because this lipid does not easily exchange, spontaneous lipid exchange to the apparent uptake appears unlikely.
After liposome uptake, more than 80% of the intracellular label from the DPPC was found in the PC fraction (table 3). This finding is comparable to those from other reports.25,28 The tendency towards increasing internalisation with increasing duration of NO2 exposure suggests an increased demand of PC in lung injury. This interpretation is in line with the observation of increased PC synthesis in type II cells after oxidant injury,10,12,13 a mechanism which enables the lung to cope with increased demand of PC in stress situations.
Lipids that have been internalised by control cells in the presence of SP-A are resecreted very rapidly at first, reaching a resecretion maximum about 20 minutes after the start of incubation. This time point allows a good comparison between all experimental groups. Cells from NO2 exposed lungs did not show such a clear maximum resecretion in the resecretion kinetics studies (fig 4). In these cells more label was retained in the exposed cells than in control cells, confirmed by the lipid material found after resecretion in the media (fig 5). As in the uptake experiments, no difference was found for the resecretion of lipid that was internalised in the absence and presence of SP-A in the different NO2 exposure groups. The most striking argument for lipid internalisation, however, is the occurrence of radioactivity in the lamellar bodies (table 2).
The finding that type II cells from NO2 exposed lungs internalised more lipid but resecreted less than the cells from control lungs suggests that internalised lipid is either stored or metabolised and used for de novo synthesis of PC. The fact that the resecreted lipid shows a clear decrease in PC label favours metabolism. This decrease shows a tendency with the duration of exposure to NO2. On the other hand, the phosphatidylglycerol contained in the liposomes has a stimulating effect on the rate of PC synthesis that has been shown through activation of the rate limiting enzyme of the pathway, choline phosphate cytidylyltransferase.29
To date, treatment with surfactant has been used mostly in IRDS and in a few cases of ARDS.30,31 Because surfactant is also impaired in chronic obstructive pulmonary disease (COPD), it is possible that such patients might benefit from treatment with surfactant and recent studies have described its use in adult patients with bronchitis and bronchiolitis.32,33 These studies clearly show improvement in lung function based on replacement of structurally and functionally impaired alveolar surfactant. No information on liposome uptake and resecretion in type II cells from injured lungs has been found. This was attempted in the present experimental study. Lung injury represents a stress situation which consumes energy in which intracellular surfactant pools are limited from de novo synthesis. Uptake of surfactant-like liposomes by type II cells and resecretion of lipid on demand could cope with this situation. Our study shows for the first time that type II cells from injured lungs internalise more lipid than cells from control lungs. This internalisation could be used to take up pharmacologically active substances given to treat lung diseases, enabling the cells to direct the lipid material to intracellular pools for resecretion on demand and/or via multivesicular bodies and lysosomes to be used for de novo synthesis of surfactant phospholipid. This hypothesis is supported by other studies showing increased phospholipid synthesis in type II cells of diseased lungs.10,12,13 Moreover, if liposomes carrying drugs for treatment of lung injury were internalised, this would enable the lung to be treated directly at the place of impairment.
In summary, our study has shown for the first time that liposome uptake in type II cells from diseased lung is increased, allowing the lung to form intracellular phospholipid pools that enable it to cope with stress situations such as lung injury. The reduced resecretion does not argue against this possibility, because our experimental approach only analysed the in vitro situation and did not include factors that stimulate surfactant phospholipid secretion in vivo in diseased lungs. Further studies of the mechanisms involved in surfactant activity are necessary to understand better the basic mechanisms of surfactant metabolism in type II cells of diseased lungs to develop treatment strategies for lung diseases such as COPD.
Acknowledgments
The authors thank Ms Frauke M Koerner and Mrs Annette Püchner for excellent technical assistance.
REFERENCES
Footnotes
-
The study was supported by the BMBF Grant No. 01GC6901/5, Clinical Research group “Chronic Lung Injury”.