Article Text

Abstract
Background: Previous studies have indicated the benefits of adding long acting β2 agonists to inhaled corticosteroids in the maintenance treatment of moderate to severe asthma. The effects of adding eformoterol to corticosteroids on asthma control and exacerbations in patients with mild to moderate asthma were studied.
Methods: After a run in period of 7–14 days on existing medication, 663 symptomatic patients were randomised to receive budesonide Turbohaler 400 μg twice daily together with either eformoterol Turbohaler 9 μg (delivered dose) or placebo twice daily. After 4 weeks patients whose asthma was well controlled (n=505) were re-randomised to receive budesonide 400 μg daily and either eformoterol 9 μg or placebo twice daily for a further 6 months.
Results: Patients receiving eformoterol achieved asthma control 10 days sooner than those receiving budesonide alone, and improvements in lung function, symptoms, quality of life, and relief β2 agonist use were significantly greater with eformoterol. During the 6 month follow up the frequency of mild exacerbations was significantly lower in the eformoterol group than in those receiving budesonide alone (7.2 versus 10.5 per patient, 95% confidence interval for ratio 0.49 to 0.96, p=0.03). The time to first day of poorly controlled asthma was 97 days in the eformoterol group compared with 42 days in the placebo group (p=0.003).
Conclusions: Adding eformoterol to a low or moderate dose of budesonide in mild asthma resulted in faster and more effective control than treatment with budesonide alone. Eformoterol allowed the corticosteroid dose to be reduced while also decreasing the rate of mild exacerbations compared with budesonide alone. These data suggest a therapeutic advantage of adding eformoterol to inhaled corticosteroids in patients with mild to moderate asthma.
- asthma
- eformoterol
- budesonide
- dose reduction
Statistics from Altmetric.com
The use of inhaled corticosteroids to control the inflammatory response underlying asthma is the basis for effective long term control in all but the most intermittent cases.1 Current British Thoracic Society (BTS) and Global Initiative for Asthma (GINA) guidelines emphasise the importance of gaining prompt control of asthma and its symptoms, recommending that patients with persistent asthma should initially be given a moderately high dose of inhaled corticosteroid which should be reduced when control is achieved.2,3 For patients whose symptoms are not controlled by low dose corticosteroids, recent guidelines have suggested that adding a long acting β2 agonist achieves a similar degree of control to that seen following an increase in the inhaled corticosteroid dose.2
Eformoterol (Oxis, AstraZeneca UK Ltd) is a moderately lipophilic β2 agonist with a rapid onset of action (within 1–3 minutes) and a duration of effect of at least 12 hours.4–11 A recent study in patients with moderate to severe asthma (FACET) showed that the addition of eformoterol 9 μg twice daily to either low or high dose budesonide resulted in significant reductions in mild and severe asthma exacerbations.12 However, many patients with milder asthma may also benefit from adding eformoterol to their treatment, but such patients have not been studied.
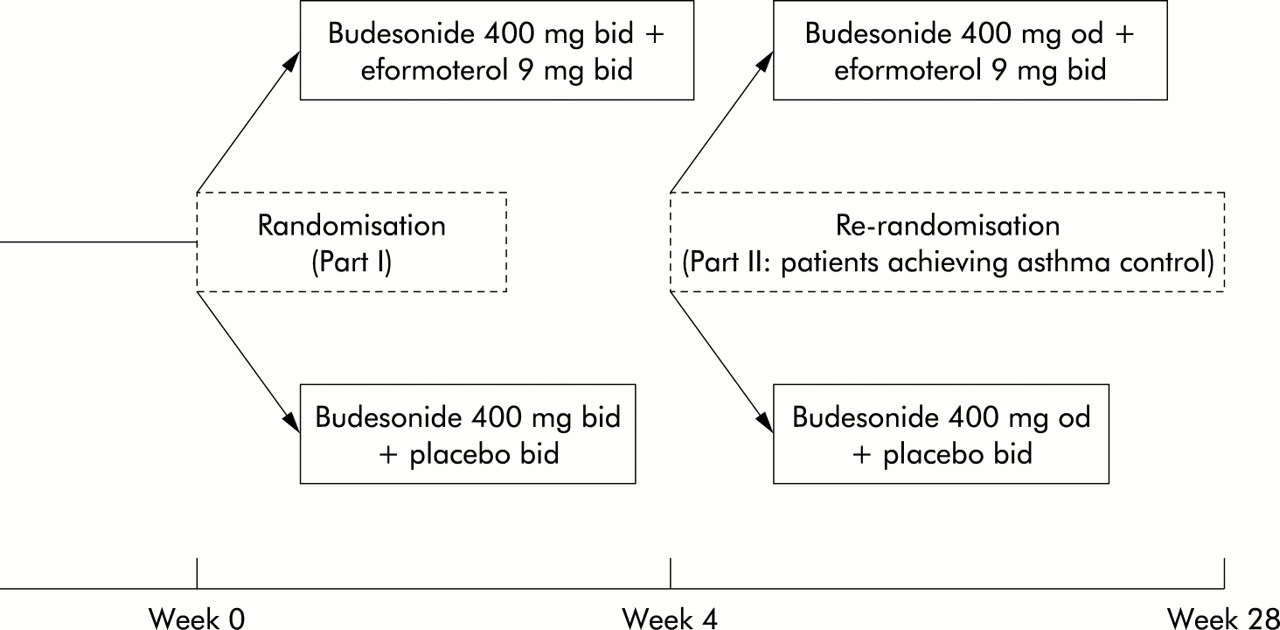
This study investigated the role of eformoterol Turbohaler in the treatment of mild to moderate symptomatic asthma; inclusion criteria were broad so the study closely resembled clinical practice. The study had two primary objectives. The primary objective in part I, which lasted 4 weeks, was to determine the effect of adding eformoterol to a moderate dose of inhaled corticosteroid (400 μg budesonide twice daily) on length of time to achieve asthma control. In part II, which lasted 6 months, the primary objective was to determine the effect of adding eformoterol to a lower dose of inhaled corticosteroid (400 μg budesonide once daily) on the time to the first mild asthma exacerbation.
METHODS
Study design
This multicentre, double blind, randomised, parallel group study was conducted in 152 general practices in the UK and Republic of Ireland, and comprised two parts (fig 1). The study was preceded by a run in period of 7–14 days to establish baseline values, during which patients continued on their pre-study medication.

Study design.
Patients
Patients aged > 12 years with a diagnosis of asthma confirmed in the clinical record for > 3 months were recruited. Current treatment had to include a short acting β2 agonist alone or with an inhaled corticosteroid (< 400 μg/day beclomethasone dipropionate or budesonide via pressurised metered dose inhaler, or < 200 μg/day fluticasone or budesonide via Turbohaler) at a constant dose for > 4 weeks. In addition, patients were required to have experienced asthma symptoms (chest tightness, cough, wheeze, or shortness of breath) on a minimum of 3 days in the week before enrolment into the study.
After baseline values had been established, patients were eligible for randomisation into part I of the study on fulfilment of the following criteria: asthma symptoms on > 3 days of the previous 7 days; either reversibility of peak expiratory flow (PEF)/forced expiratory volume in 1 second (FEV1) of > 12% (or > 9% of predicted normal), or a diurnal variation of > 20% on at least one day during the run in period.
Patients with more severe or recently unstable asthma were excluded: PEF <50% predicted; currently receiving (during 4 weeks before enrolment) nebulised therapy, oral corticosteroids, leukotriene antagonist, or long acting β2 agonist; a clinically relevant upper respiratory tract infection in the 4 weeks leading up to enrolment. Patients with irreversible chronic airways disease were also excluded.
Multicentre and local research ethics committee approval was obtained for the study. All patients (and parent/guardian where necessary) gave informed written consent before any study procedures were carried out. The study was conducted in accordance with the principles of Good Clinical Practice.
Study treatment
Patients who were eligible for part I were randomly assigned to one of two treatment arms for 4 weeks: budesonide 400 μg (Pulmicort Turbohaler, AstraZeneca UK Ltd) and eformoterol 9 μg (Oxis Turbohaler, AstraZeneca UK Ltd), both twice daily (referred to as BUD800+EF); or budesonide 400 μg and placebo Turbohaler, both twice daily (referred to as BUD800+PL). Patients continued to use their normal relief medication (short acting β2 agonists) as needed.
Patients with stable asthma after part I of the study entered part II. Asthma was defined as stable if none of the following had occurred during part I: diurnal variation of >20% in PEF on two consecutive days; use of > 4 inhalations of β2 agonist per day on two consecutive days; reduction in PEF >30% baseline; awakening due to asthma on two consecutive nights; or the need to use oral glucocorticoids.12 Patients were re-randomised to receive a further 6 months of treatment with either budesonide 400 μg at night—that is, half the dose of inhaled corticosteroid—and eformoterol 9 μg twice daily (BUD400+EF) or budesonide 400 μg at night and placebo twice daily (BUD400+PL). As in part I, patients continued to use their normal relief medication.
Treatments prohibited during the study were astemizole, oxitropium bromide, ipratropium bromide, and theophylline. Introduction of regular treatment or changes in doses of nasal corticosteroids, antihistamines, or anti-inflammatory treatments for asthma were also prohibited.
Efficacy assessments
There were six scheduled visits to the clinic: at the start of the run in and treatment periods and after 1, 3, 5, and 7 months of treatment. In addition, telephone contacts were scheduled after 4 and 6 months of treatment. At scheduled clinic visits, patients graded their daytime and night time asthma symptoms (cough, wheeze, chest tightness, and shortness of breath) on a 4 point scale (0=no symptoms, 1=symptoms, but not affecting any activities during the day/sleep at night, 2=symptoms affecting at least one daily activity or disturbing sleep, and 3=symptoms affecting > 2 daily activities or disturbing sleep all night or most of the night). PEF was recorded at each clinic visit. The occurrence of adverse events was assessed using a standard question.
Patients completed a diary card every morning and evening during the run in and treatment periods, recording PEF before asthma medication, relief β2 agonist usage, and grade of daytime and night time symptoms (according to the 4 point scale described above). Sleep disturbance was derived from the night time symptom score—a score of 2 or 3 represented a night of disturbed sleep. Compliance with use of eformoterol was assessed on a weekly basis, with patients recording the number of missed doses. Days off work or school because of asthma were also recorded.
Patients completed the self-administered Mini Asthma Quality of Life questionnaire13 on their own, at clinic visits at the start of the run in and treatment periods, and at the end of parts I (4 weeks) and II (7 months). A clinically relevant change in quality of life (QoL) was defined as a change in overall score of >0.5.14
Primary outcome measures
The primary outcome measure in part I was time to asthma control—that is, three consecutive days with a symptom score of 0 based on diary card daytime and night time symptom assessments. In part II the primary outcome measure was time to the first mild asthma exacerbation.
During part I a mild exacerbation was defined as in the FACET study.12 As only stable patients were randomised into part II of the present study, a mild exacerbation was redefined as any combination of the following on two consecutive days: PEF < 80% of baseline values, β2 agonist use of > 4 inhalations above baseline on completion of part I, or awakenings during the night because of asthma.
Secondary outcome measures
Secondary outcomes were time to first severe asthma exacerbation, frequency of mild and severe exacerbations, and proportion of patients free of exacerbations during 6 months of treatment. A severe exacerbation was defined in both parts I and II as requiring oral corticosteroid treatment or as a decrease in morning/evening PEF >30% of baseline on two consecutive days. A maximum of three severe exacerbations requiring additional treatment was allowed during the whole study. Patients exceeding these criteria were withdrawn.
In part II the time to the first poorly controlled day and the frequency of poorly controlled days were determined. A poorly controlled day was defined as one involving any of the following: PEF < 80% of baseline values, β2 agonist use > 4 inhalations above baseline, or awakenings during the night because of asthma.
In addition, changes in QoL and differences in the nature and frequency of adverse events between treatments were evaluated.
Sample size
It was planned to randomise 600 patients into part I, of whom 450 were required to continue into part II. With 663 and 505 patients randomised into parts I and II, respectively, the study had a power of >80% to detect the pre-specified clinically significant differences of 2 days in the median time to asthma control and 30 days in the median time to first mild exacerbation.
Statistical analysis
Efficacy was analysed using an intent-to-treat approach using all available data. Survival analysis techniques were used to analyse median times to asthma control in part I and median time to first mild exacerbation in part II. Data were summarised by the median time (where applicable) and the Kaplan-Meier survival estimates at day 28 (part I) and day 168 (part II).
Time to first severe exacerbation and first poorly controlled day were compared between treatments using the log rank test. Cox regression was used to investigate the association of previous inhaled corticosteroid use and treatment during part I with time to asthma control in part I and time to mild and severe exacerbations in part II. Poisson regression was applied to investigate the influence of previous inhaled corticosteroid use and treatment in part I on the frequency of mild and severe exacerbations in part II. For the diary card assessments, mean values for the 7 days before each visit were calculated. Daily diary card assessments, number of days off work or school, and the QoL questionnaire were analysed between treatments by the Wilcoxon rank sum test and within treatments by the Wilcoxon signed rank test. The frequency of mild exacerbations, severe exacerbations, and poorly controlled days were analysed for treatment effects by a Poisson regression model, adjusted for duration of treatment during part II. Adverse events were summarised by descriptive statistics and any apparent difference between groups was investigated by a χ2 test. The data were analysed using SAS for Windows version 6.12. Exact p values for χ2 tests were calculated using statxact version 2.11.
Treatment effects are presented as either the difference between eformoterol and placebo or the ratio of eformoterol to placebo.
RESULTS
Study population
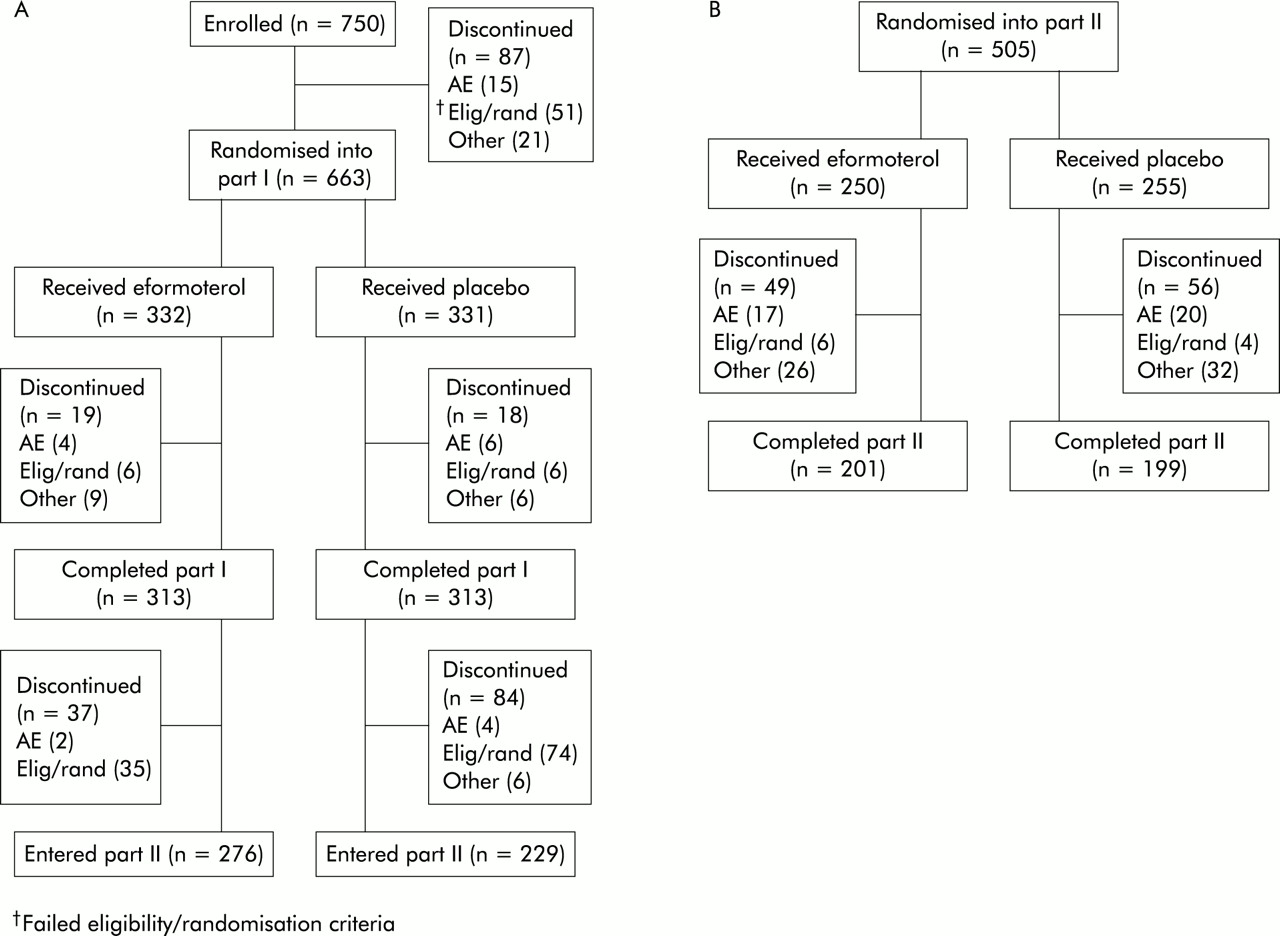
A total of 750 patients were enrolled into the study, 87 of whom discontinued the study before randomisation into part I; 15 withdrew because of an adverse event, 51 failed the eligibility/randomisation criteria, and 21 discontinued for other reasons. Stated compliance during parts I and II appeared high, with 99% of patients in both groups reporting >75% compliant days throughout the whole study period. Of the 663 patients randomised into part I, 332 received BUD800+EF and 331 received BUD800+PL. A total of 626 patients completed part I and 505 were randomised into part II, with 250 receiving BUD400+EF and 255 receiving BUD400+PL. The main reason for discontinuation was failure to meet the eligibility/randomisation criteria. The treatment groups were comparable with respect to patient characteristics on entry to either part of the study (table 1). Patient disposition in the two parts of the study is summarised in fig 2.
Mean (SD) demographic characteristics at entry

Patient disposition: (A) part I; (B) part II.
Part I
Primary outcome measures
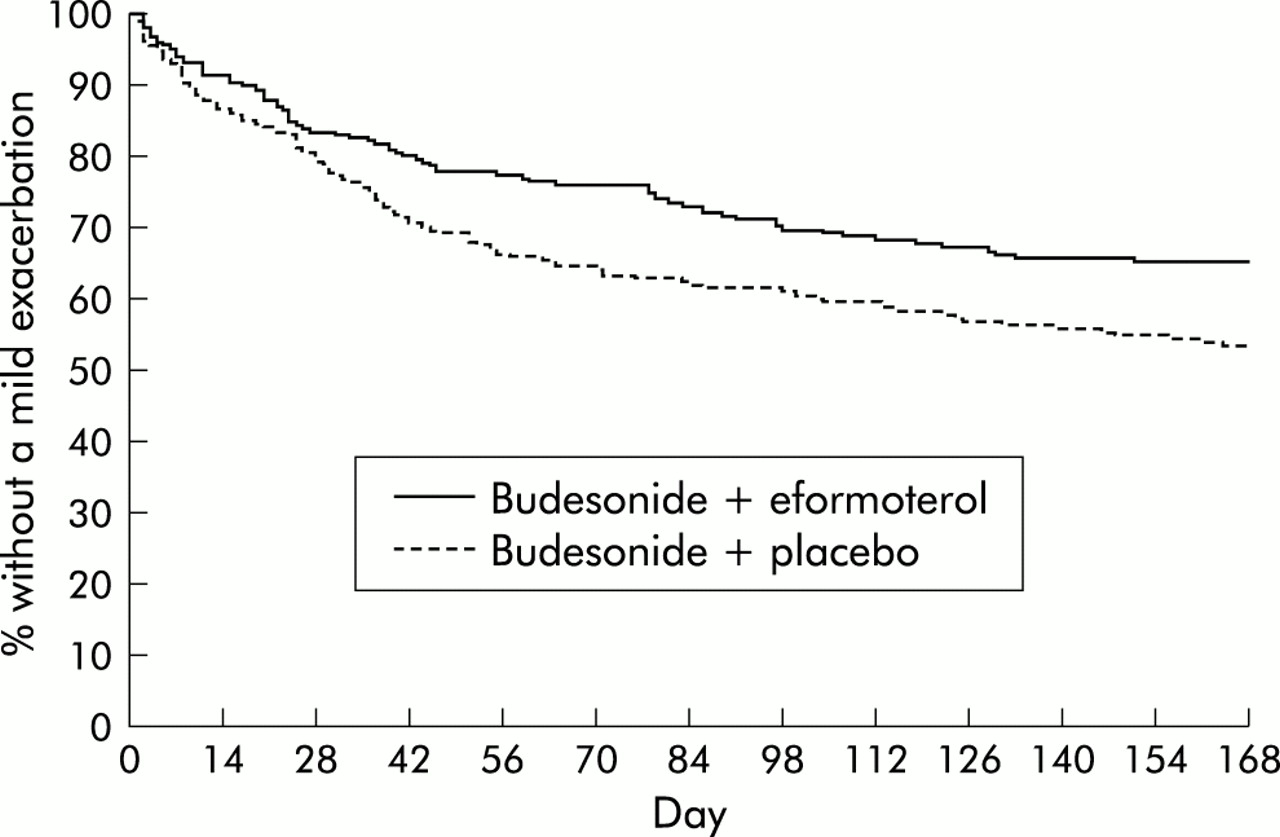
Median time to symptom relief was significantly shorter for patients receiving BUD800+EF than for patients receiving BUD800+PL (24 v 34 days; p=0.003). A significantly greater proportion of patients in the eformoterol group achieved asthma control at 4 weeks (52% v 41%, difference 11%; 95% confidence interval (CI) 3 to 19; p=0.003). The survival analysis provided evidence that the time to first mild exacerbation was significantly longer in the BUD400+EF patients than in the BUD400+PL patients; an estimated 35% and 47% of patients, respectively, experienced a mild exacerbation by 24 weeks (difference –12%; 95% CI –21 to –3; p=0.01, fig 3).

Survival analysis of time to first mild exacerbation during part II of study.
Secondary outcome measures
Relief β2 agonist use and symptom free days
Both treatment groups showed a statistically significant reduction in daytime relief β2 agonist use after 4 weeks of treatment compared with baseline (p<0.001; table 2, fig 4). This reduction was significantly greater with BUD800+EF than with BUD800+PL (–1.18 v –0.85 inhalations/day, difference –0.34, 95% CI –0.55 to –0.13; p<0.001). Moreover, the BUD800+EF group experienced more symptom free days (9.7 v 7.5 days, difference 2.2, 95% CI 0.8 to 3.6; p=0.004) and symptom free days with no relief inhaler use (7.6 v 5.7 days, difference 1.9, 95% CI 0.6 to 3.2; p=0.01) than the placebo group.
Mean (SD) run in response and mean change in daily diary card assessments of daytime and night time reliever inhaler use, symptom severity, and nocturnal disturbance. Minimum and maximum values are shown in square brackets

Daytime β2 agonist use during the last 7 days of the run in period and days 1–28 of part I.
Lung function
At 4 weeks both treatment groups had significant (p<0.001) increases in diary PEF compared with baseline (figs 5 and 6). However, the mean change from run in to 4 weeks in both morning and evening PEF was significantly greater for patients receiving BUD800+EF than for patients receiving BUD800+PL (morning +36.8 v +17.8 l/min, difference 19.0, 95% CI 12.3 to 25.6, p<0.001; evening +26.0 v +10.2 l/min, difference 15.7, 95% CI 9.4 to 22.1, p<0.001).
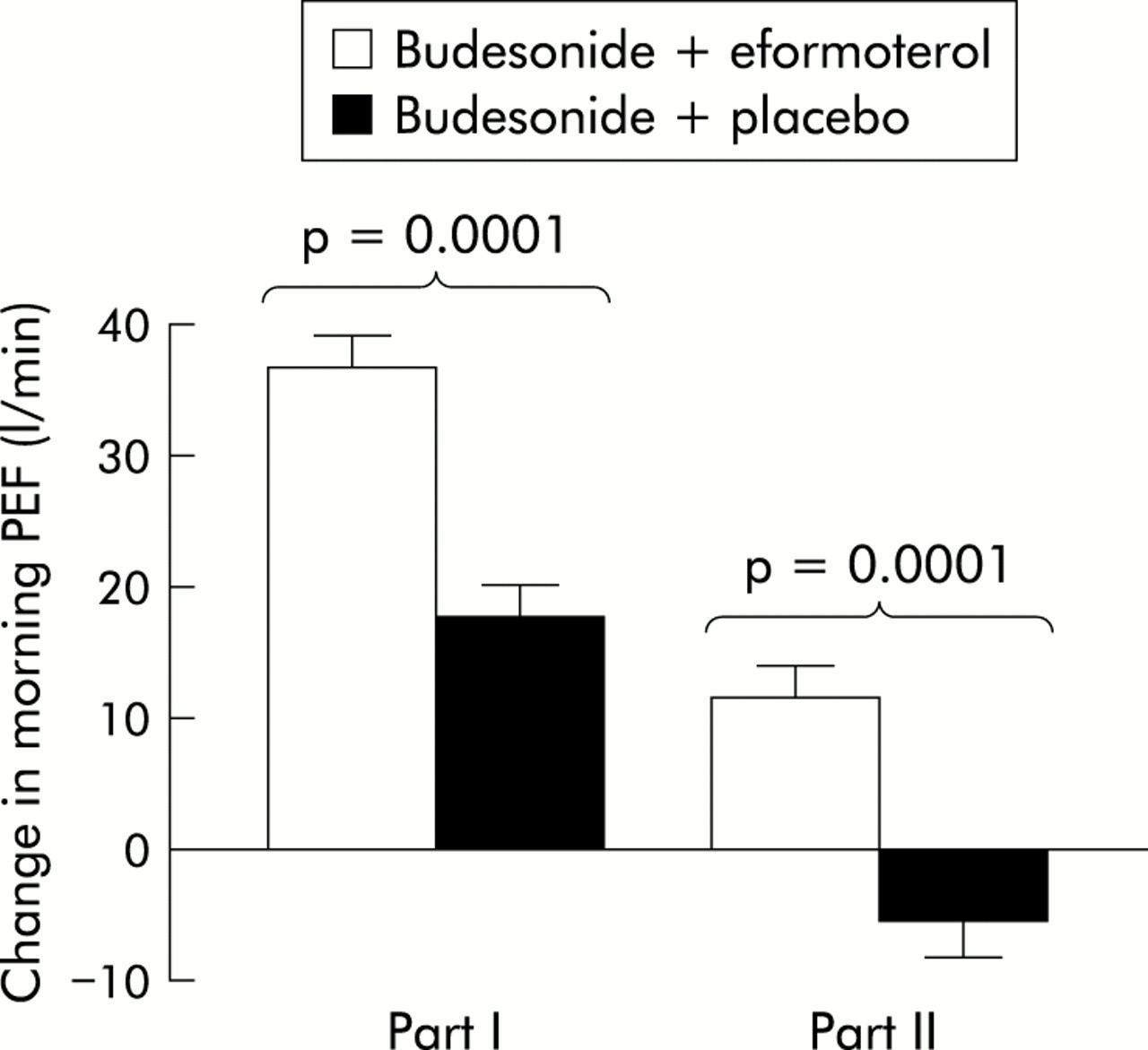
Mean (SE) change from baseline in morning PEF (p values between treatments); part II data relate to the change after 6 months.

Mean (SE) change from baseline in evening PEF (p values between treatments); part II data relate to the change after 6 months.
Asthma symptoms
Significant improvements were observed in all daily diary card assessments after 4 weeks of treatment within both treatment groups (all p<0.001). However, with the exception of night time symptom severity, patients receiving BUD800+EF experienced significantly greater improvements in each parameter than those receiving BUD800+PL (table 2).
There was no significant difference between treatment groups in the percentage of days taken off work or school because of asthma in part I of the study.
Quality of life
Clinically relevant improvements in the overall QoL score were achieved by 51% of patients receiving BUD800+EF and 47% of patients receiving BUD800+PL. The mean improvement was significantly greater with BUD800+EF than with BUD800+PL (0.67 v 0.48, difference 0.19, 95% CI 0.04 to 0.33; p=0.04, fig 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (SE) change in overall quality of life (QoL) score from baseline.
Part II
Primary outcome measures
Fewer than 50% of patients in each group experienced a mild exacerbation by the assessment time point; the median time to first mild exacerbation therefore could not be estimated.
Secondary outcome measures
Asthma exacerbations
Addition of eformoterol to budesonide led to a significantly lower estimated frequency of mild exacerbations per patient compared with the placebo group (7.2 v 10.5 per 6 months, frequency ratio 0.69, 95% CI 0.49 to 0.96; p=0.03). No significant differences in the frequency of severe exacerbations, which was low in both groups, or time to first severe exacerbation were observed between the groups.
Poorly controlled days
The median time to first poorly controlled day was more than halved by addition of eformoterol to budesonide (from 97 to 42 days) with an estimated 58% and 69% of patients experiencing a poorly controlled day by 6 months (difference –11%, 95% CI –20 to –2; p=0.003). The estimated frequency of poorly controlled days was also significantly less for patients receiving BUD400+EF than BUD400+PL (10.0 v 14.2 days per patient per 6 months, frequency ratio 0.70, 95% CI 0.52 to 0.95; p=0.02).
Relief β2 agonist use and symptom free days
Relief β2 agonist use (day and night) was significantly lower in the BUD400+EF group than in the BUD400+PL group at 2 months (p<0.001 (daytime) and p=0.02 (night time)), 4 months (p=0.002, day and night) and 6 months (p<0.001, day and night). Furthermore, patients receiving BUD400+EF benefited from an extra 17.4 symptom free days (89.0 v 71.6 days, difference 17.4, 95% CI 6.4 to 28.7; p=0.002) and 20.3 symptom free days with no relief inhaler use (77.4 v 57.1 days, difference 20.3, 95% CI 9.4 to 31.4; p<0.001) compared with BUD400+PL.
Lung function
Significant improvements in morning and evening PEF were observed in the eformoterol treated group despite a halving of their corticosteroid dose. Between treatments significant differences in the changes in PEF were observed in favour of BUD400+EF after 6 months (morning: difference 17.1 l/min, 95% CI 9.7 to 24.6, p<0.001; evening: difference 17.3 l/min, 95% CI 9.6 to 25.0, p<0.001). The differences seen at 2 and 4 months were similar and also significant.
Asthma symptoms
After a further 6 months of treatment significant changes in diary card responses were observed in favour of the BUD400+EF group for all parameters except sleep disturbance (table 2). As only stable patients entered part II of the study, data from the final week of part I were used as baseline so that changes reflected only those observed during part II. There was no significant difference between treatment groups in the percentage of days taken off work or school because of asthma in part II of the study.
Quality of life
At the end of part II patients treated with BUD400+EF reported further improvements in overall QoL score (mean improvement +0.23, p<0.001). Neither the change within the BUD400+PL group (+0.03, p=0.2) nor the difference between treatments (0.20, p=0.1) was statistically significant.
Influence of previous corticosteroid use
On entering part I of the study, 446 patients had already been treated with inhaled corticosteroids and 217 had just started treatment with inhaled corticosteroids. There was no evidence that previous corticosteroid use had any influence on any of the outcome measures (table 3).
Influence of previous steroid use on the time to achieve asthma control, time to first mild or severe asthma exacerbation, and frequency of mild and severe exacerbations
Association between treatment during part I and outcome during part II
The time to first mild or severe exacerbation and the frequency of exacerbations during part II were not influenced by the treatment received in part I. The most favourable outcomes in terms of PEF, relief inhaler use, symptom score, and sleep disturbance were observed in patients who received eformoterol in both parts of the study (table 4). After gaining control on their first treatment regimen, there was a tendency for patients switching from budesonide alone to low dose budesonide plus eformoterol to show further improvements and a tendency for patients switching from budesonide plus eformoterol to low dose budesonide alone to get slightly worse. The other two subgroups—that is, patients reducing their steroid dose only—tended to remain stable. These trends are to be expected, and are also reflected in the mild exacerbation rates observed during part II (BUD800/BUD400+EF 31%, BUD800+EF/BUD400 50%, BUD800+EF/BUD400+EF 38%, BUD800/BUD400 43%).
Mean (SD) changes in all daily diary card assessments from run in to 6 months. Minimum and maximum values are shown in square brackets
Safety
The safety profiles observed in both phases of the study were similar across treatment groups. The most common adverse events were headaches and respiratory system disorders (infections and worsening asthma).
In part I of the study more patients taking budesonide alone experienced a deterioration in asthma compared with those taking eformoterol (6% v 1.2%, p<0.001), which could partly be explained by the increased number of respiratory infections in the BUD800+PL group (3.3% v <1%, p=0.003). The proportion of patients reporting worsening asthma during part II was similar for both groups.
DISCUSSION
This study investigated the use of the long acting β2 agonist eformoterol to achieve and maintain asthma control in patients with mild to moderate symptomatic asthma. After 4 weeks of treatment there were significant improvements in both the budesonide alone and budesonide plus eformoterol groups, indicating that symptom control can be achieved with inhaled corticosteroid alone. However, treatment with budesonide and eformoterol provided a clear therapeutic advantage over treatment with budesonide alone, enabling asthma control to be achieved more quickly and in a greater proportion of patients, regardless of whether or not they had been receiving inhaled corticosteroids before the study. In part II, when asthma control was achieved and the dose of budesonide was halved, combined treatment also showed clear benefits over budesonide alone. Over a 6 month period the rate and frequency of mild exacerbations was significantly reduced in those patients receiving eformoterol compared with those patients receiving corticosteroid only; very few patients in either group experienced a severe exacerbation, and the first poorly controlled day occurred a median of 55 days later with additional eformoterol treatment than with budesonide alone.
The 12 month FACET study conducted in patients with moderate to severe asthma showed clear benefits of adding a long acting β2 agonist to a corticosteroid in terms of a significant reduction in mild and severe exacerbations and improvement in lung function.12 The present study has extended these findings and has shown that similar benefits can be achieved by adding eformoterol to the treatment regimens of patients with milder asthma. As in the FACET study, asthma control in our study was also significantly better with eformoterol than with placebo after the dose of budesonide was “stepped down” in patients whose asthma was stabilised.
The aim of an asthma treatment strategy is to gain prompt control of asthma symptoms. The median time to achieve asthma control in part I of this study occurred 10 days earlier when eformoterol was added to budesonide compared with the addition of placebo (24 v 34 days). A very tight definition of asthma control was used in this study, but a “real life” acceptable level of symptoms for patients may be less demanding than this, and asthma control may therefore be attained even more quickly in clinical practice. The addition of eformoterol resulted in a significantly greater reduction in short acting β2 agonist use from as early as the second day and significantly greater improvements in morning and evening PEF from the start of treatment to the end of the fourth week of treatment. Importantly, the observed improvements seen at 4 weeks of eformoterol treatment were evident throughout the 4 week treatment period, indicating the very rapid response to the eformoterol treatment regimen.
In part II of the study administration of the reduced dose of inhaled corticosteroid alone resulted in 47% of patients experiencing a mild exacerbation over the 6 month period. In contrast, the addition of eformoterol was associated with a lower rate (35%, p=0.01). The estimated frequency of mild exacerbations over a 12 month period was lower in this study than in the FACET study (14.4 v 21.3 BUD400+EF, 21 v 35.4 BUD400+PL), probably because of the different populations being investigated (mild/moderate asthma v moderate/severe asthma). Very few patients suffered a severe exacerbation in either study.
Significant additional benefits (both on asthma symptoms and lung function) were observed in both parts of the study when eformoterol was added to budesonide. This finding suggests that eformoterol has two independent roles: firstly, to gain effective symptom control rapidly (in conjunction with a moderate dose of corticosteroid) and, secondly, to maintain asthma control when the dose of inhaled corticosteroid has been reduced. Additional analyses indicated that the outcomes observed during part II were not dependent on the treatment received in part I of the study. Patients who received eformoterol in both study phases had the best outcomes in terms of PEF, relief inhaler use, sleep disturbances, and symptom scores. The greater control of asthma observed with the use of eformoterol was translated into an improved QoL for patients. In part I a significantly greater improvement in QoL score was observed in the BUD800+EF group than in the BUD800+PL group. The improvement in QoL was sustained following re-randomisation into part II, despite the dose of budesonide being reduced. No safety issues were identified when eformoterol was added to budesonide.
This study has shown that adding eformoterol to a moderate dose of inhaled budesonide gave earlier asthma control than with placebo. In patients whose asthma was brought under control, the addition of eformoterol significantly reduced mild exacerbations compared with placebo when the dose of inhaled budesonide was reduced. The FACET study showed the therapeutic advantage of eformoterol in patients with moderate to severe asthma; the present study has shown that this therapeutic advantage also extends to patients with mild to moderate asthma. In combination, these studies indicate that addition of eformoterol to inhaled budesonide will benefit a broad spectrum of patients with asthma.
Acknowledgments
The authors thank J Haughney, D Bellamy, and LM Campbell for valuable advice on the study design, Emma Beresford who performed the statistical analyses, and all the general practitioners in the UK and Republic of Ireland who contributed to the study as the FLOW Research Group. Financial support for this study was provided by AstraZeneca UK Ltd.