Article Text

Abstract
The prevalence, prognosis, clinical presentation, pathophysiology, diagnosis, and treatment of the central sleep apnoea syndrome (CSAS) are reviewed and its relationship with congestive heart failure (CHF) is discussed. Adequately powered trials are needed with survival and health status as end points to establish whether correction of sleep related breathing abnormalities improves the outcome in patients with CHF.
- central sleep apnoea syndrome
- Cheyne-Stokes respiration
- congestive heart failure
- periodic breathing
Statistics from Altmetric.com
In recent years a number of studies have been published investigating the relationship between chronic congestive heart failure (CHF) and the central sleep apnoea syndrome (CSAS). CSAS has a hypercapnic and a hypocapnic form. The hypercapnic form results from a chronically depressed respiratory drive or ability to breathe, and is associated with hypercapnia during wakefulness and sleep.1 Hypocapnic CSAS, which is discussed in this paper, is much more prevalent. It is not caused by snoring or mechanical obstruction of the upper airway (obstructive sleep apnoea syndrome), but has a completely different cause and pathophysiology (fig 1).2

Taxonomy of the central sleep apnoea syndrome.
The typical nocturnal breathing pattern in the majority of patients with hypocapnic CSAS is of periodic breathing, characterised by a regular, crescendo-decrescendo oscillation of tidal volume (fig 2) which is thought to be caused by dysfunction of central respiratory control. As the central drive to breathe slowly fades, ventilation temporarily ceases before resuming again. This results in an oscillation between central hypopnoea (a decrease of ≥50% in the sum of thoracoabdominal movements lasting 10 seconds or more followed by a decrease of ≥4% in peripheral oxygen saturation (Sao2)) or central apnoea (a reduction of >90% in thoracoabdominal movement or complete cessation of ventilatory efforts) and hyperventilation.3 The typical length of one period is 30–60 s, but longer periods have been observed. During a prolonged hypopnoea or apnoea the oxygen saturation decreases, combined with a slow rise in the arterial carbon dioxide tension (Paco2) and a decline in blood and tissue pH. Central nervous system activity, derived from cortical electroencephalography (EEG), in most cases remains unaffected until late apnoea. However, changes towards lower activity and deeper sleep stages during apnoea have occasionally been found.4 In many cases the resumption of ventilation is associated with EEG arousals, which are markers of central nervous system activation,5 and disturbed sleep which is driven back to a more superficial level.

Five minute section of a polysomnographic record of EEG (C3A1, C4A2), electrocardiogram (ECG), oronasal air flow (Flow), abdominal ventilatory effort (ABD), thoracic ventilatory effort (THO), and oxygen saturation measured at the finger tip of the left hand (Sao2) in a 57 year old man with moderate ischaemic CHF (NYHA II–III). Typical breathing pattern with Cheyne-Stokes respiration with hyperpnoeic and apnoeic sequences in sleep stage 2. There are fluctuations in oxygen saturation in response to periodic breathing, with delay of the transit time from the lungs to the finger tip of the left hand.
Cheyne-Stokes respiration (CSR) is commonly used as a synonym for periodic breathing, but some authors distinguish between the two. If the intervals of hyperventilation are separated by hypopnoeas only it is described as periodic breathing, but if both hypopnoeas and apnoeas are present it is termed CSR (fig 2).6,7 In this review, hypocapnic CSAS and periodic breathing are used as synonyms. It had been suggested that three or more successive periods are necessary for the diagnosis of periodic breathing,8 but there is no generally accepted definition.
In most patients periodic breathing is present during light sleep stages 1 and 2 and rarely during rapid eye movement (REM) sleep. The amount of periodic breathing varies widely between different patients, but seems to be constant over successive nights in an individual.9
PREVALENCE
Although found in a number of diseases and in some normal subjects, particularly at high altitude, the highest prevalence of CSAS is found in patients with severe chronic left ventricular insufficiency, in whom it was first described by Cheyne in 181810 and again by Stokes in 1854.11 Recent reports12,13 estimate the prevalence of periodic breathing in patients with underlying ischaemic heart disease or idiopathic dilated cardiomyopathy, a maximum ejection fraction of 40%, and optimised medical treatment to be 45–50%. Most of these patients are male. Whether this reflects the unequal sex prevalence of cardiac diseases or whether sex hormones play an additional role is not known.14
CHARACTERISTICS AND PROGNOSIS IN HEART FAILURE
The presence of periodic breathing during sleep in patients with CHF has significant implications. Patients with CHF and periodic breathing are more limited in their physical performance and develop dyspnoea at lower workloads than patients with disease of similar severity but without periodic breathing.15 On average, the left ventricular ejection fraction is lower in patients with periodic breathing and the prevalence of cardiac arrhythmia is significantly higher than in patients with the same degree of heart failure but without periodic breathing.16,17
The prognosis of patients is worse if CHF is associated with a sleep apnoea syndrome.18 Hanly and co-workers19 conducted a survey of patients in chronic CHF with and without nocturnal CSR matched for age, sex, body mass index, severity and duration of heart disease, and cardiac medication. The cumulative survival and transplant free rate was significantly worse for patients with CSR (100% v 66% after 1 year, 86% v 56% after 2 years, respectively; fig 3). The apnoea-hypopnoea index (see below) has been shown to be an independent predictor of poor prognosis.20 Andreas and co-workers8 found an increased likelihood of dying within a few months in patients with CHF and CSR during wakefulness. Complex arrhythmias, including (non-sustained) ventricular tachycardia, are much more prevalent in patients with CSR.21
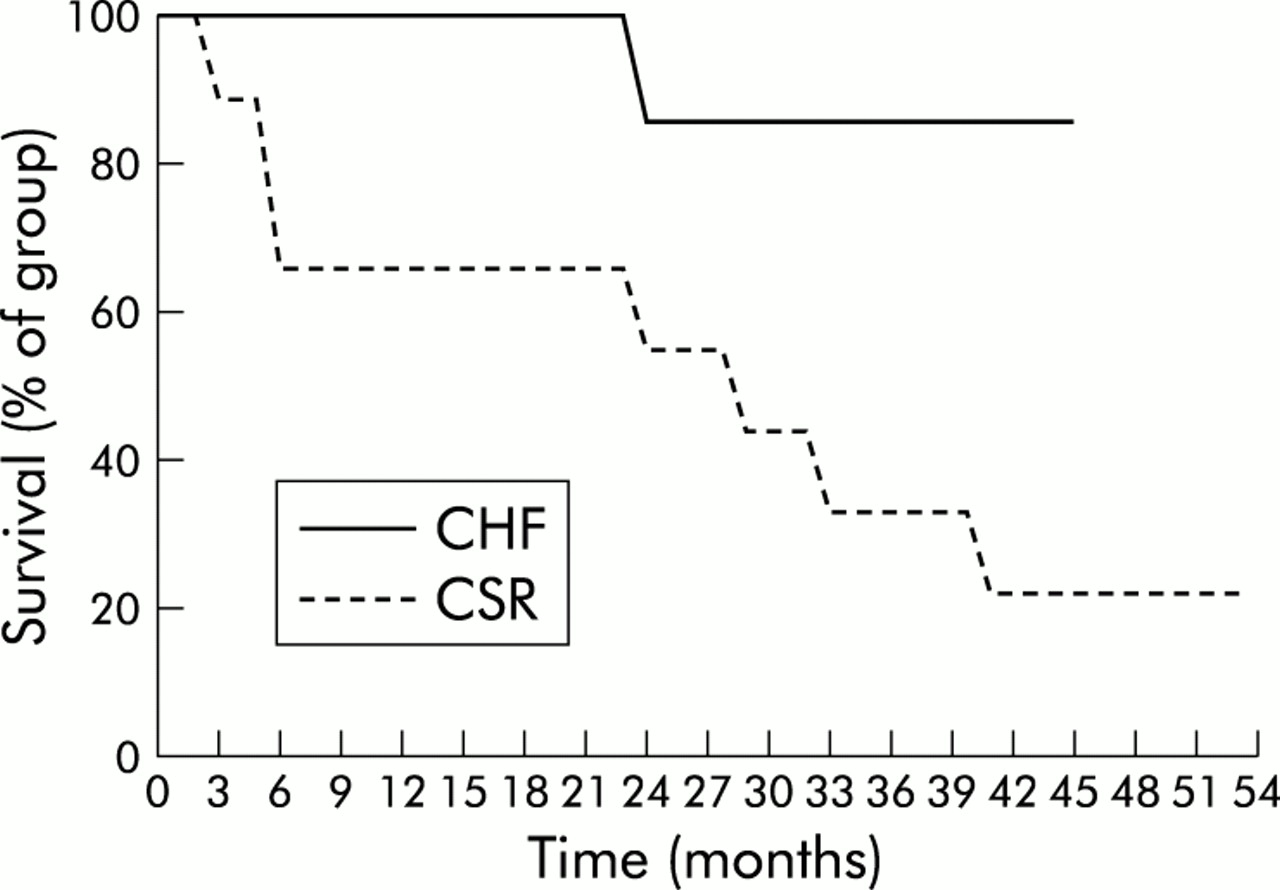
Cumulative survival of patients in chronic heart failure (CHF) with or without Cheyne-Stokes respiration (CSR). Reproduced with permission from Hanly and Zuberi-Khokhar.19
CLINICAL PRESENTATION
The typical symptoms of CSAS are daytime hypersomnolence and fatigue. Repetitive arousals cause sleep fragmentation with a reduction in the amount of slow wave and REM sleep which are the most refreshing sleep stages.22,23 Artificial sleep fragmentation in volunteers has been shown to result in excessive daytime hypersomnolence, similar to the symptoms of patients suffering from CSAS.24
Many patients are likely to fall asleep in quiet or monotonous situations or whenever they are not occupied during the day. Despite apparently sleeping for many hours during the night, patients with CSAS may not feel fully refreshed. These symptoms impair patients' quality of life and may contribute to their sedentary life style.25 However, the symptoms of CHF such as lethargy, impaired exercise capacity, and paroxysmal nocturnal dyspnoea26 may overlap with the symptoms of the sleep apnoea syndrome.27 Fatigue can also be a side effect of cardiac medication—for example, beta blockers—but hypersomnolence and excessive lethargy should alert the clinician to the possibility of CSAS.
PATHOPHYSIOLOGY OF PERIODIC BREATHING
In healthy subjects the tidal volume and frequency of breathing (minute ventilation) is principally controlled by the arterial carbon dioxide tension (Paco2), blood pH, and the arterial oxygen tension (Pao2).28 These parameters are sensed in the arterial blood by peripheral chemoreceptors located in the carotid bodies and the aortic arch and are thought to react rapidly to short term changes in them.29 The central chemoreceptors are probably located at the ventral surface of the medulla oblongata, behind the blood brain barrier, which delays responses to changes in arterial blood gas tensions. It has been hypothesised that the central chemoreceptors determine the long term target range of blood gases.30 Information from these chemoreceptors is transferred to and processed in the respiratory centre in the brain stem, adjacent to centres which control the cardiovascular system. The relationship of the respiratory centre to other brain centres which might also influence the control of breathing—for example, circadian rhythm, sleep-wake rhythm, sleep stage, voluntary influence—is still being investigated.31 Further inputs into the respiratory centre come from pulmonary J receptors which measure stretch in the airways and the lungs, and from proprioceptors in the respiratory muscles of the diaphragm and the chest wall. Output from the respiratory centre is conducted by efferent nerves to the respiratory muscles—the “ventilatory pump”.32,33
The respiratory centre maintains blood gas tensions within a tight range according to the metabolic demands of the body. Periodic breathing reflects uncompensated instability of the feedback control of ventilation. In general, feedback control systems destabilise if their damping capacity or if other mechanisms of keeping the balance are overridden. This can occur if information transfer to the controller is delayed or controller gain is altered.34,35
Delayed information transfer
Patients with left ventricular dysfunction and low cardiac output typically have a prolonged circulation time. Pryor36 speculated about 50 years ago that this might lead to a time delay between changes in blood gas tensions in the lung and their detection in the central nervous system, adversely affecting the control of ventilation. However, several recent studies have not been able to show a predisposition or statistical association between low cardiac output and periodic breathing in patients with CHF.14,19
Increased controller gain
Increased controller gain occurs if the sensitivity of chemoreceptors is increased.37 Various hormones and drugs can alter the human chemoreceptor sensitivity, including the endogenous catecholamines noradrenaline and adrenaline.38,39 Levels of both hormones are increased in the blood and urine of patients with heart failure, probably as compensation for cardiac pump failure.40 Increased circulating concentrations of these catecholamines might increase the responsiveness of the respiratory controller to carbon dioxide, leading to hyperventilation.41,42 Latent hyperventilation during wakefulness is a common finding in these patients and a relationship between an abnormally increased ventilatory response to carbon dioxide during the day and periodic breathing during sleep has been observed.43 Their Paco2 is typically in the lower normal range or even below, keeping ventilation closer to the threshold of apnoea. Javaheri44 showed that patients with CHF and CSAS had a significantly greater sensitivity to carbon dioxide (by a factor of 2.3–3.5) than patients with CHF without CSAS. In a study of 20 men with stable advanced CHF he found a significant positive correlation between sensitivity to carbon dioxide during wakefulness and the number of episodes of apnoea and hypopnoea per hour of sleep. However, this statistical observation does not provide physiological evidence that a low daytime Paco2 indicates enhanced chemosensitivity.
Chemoreceptor gain can be diminished pharmacologically by benzodiazepines, dopamine, codeine, morphine, and alcohol,45,46 and codeine and morphine have been shown in small clinical studies to reduce the amount of periodic breathing in patients with heart failure.47
With apnoea or hypopnoea gas exchange in the lung is reduced, resulting in a rapid fall in oxygen and a rise in carbon dioxide to mixed venous levels. Arousals are frequently observed towards the end of these respiratory events, triggering an increased respiratory responsiveness to carbon dioxide and therefore reducing the damping of respiratory control.48 This induces an accelerating ventilatory drive producing tidal volumes and frequencies that overshoot what was required to normalise blood gases. The net effect is a reduction in Paco2 below the apnoeic threshold, thereby precipitating the apnoeic/hypopnoeic phase and thus perpetuating the cyclic periodic breathing.49 The arousal related change in the drive to breathe is likely to be very important in sustaining periodic breathing, and provides an explanation for the ventilation overshoot following apnoea and why periodic breathing is not seen in deep sleep.
Episodes of periodic breathing are only transient phenomena in many patients, whereas impaired cardiac function, delayed circulation time, and increased chemoreceptor susceptibility are present chronically. This observation lends further support for the hypothesis that other factors contribute to the control of ventilation. Periodic breathing is usually seen only during wakefulness and light sleep stages, and is markedly attenuated or disappears during REM sleep. It is closely related to a shift in sleep stages,50 confirming that other brain centres also have a role in the control of ventilation.51 The respiratory centre also gains information from pulmonary vagal afferents, transmitted by J receptors and C fibres which are activated by tissue stretching.32 Solin and co-workers52 demonstrated an inverse relationship between awake pulmonary capillary wedge pressure and awake Paco2. If pulmonary oedema activates tissue receptors, or if hyperventilation is induced by impaired oxygen transfer in the congested lung, Paco2 may fall below the apnoea threshold.53 A single deep breath is a common finding at the onset of a sequence with periodic breathing and, because during light sleep ventilation is exclusively metabolically controlled, the fall in Paco2 caused by these events takes it below the apnoeic threshold initiating periodic breathing (fig 4).54,55 Francis and co-workers56 recently performed a quantitative general analysis of the dynamic physiology governing cardiorespiratory stability in CHF. Their mathematical model was clinically well validated and revealed the increased chemoreflex gain, long lag to chemoreflex response, low ventilation, low cardiac output, large difference between alveolar and atmospheric carbon dioxide tensions, and small lung volume as critical factors for the development of oscillations in the feedback control of ventilation. However, the model cannot explain why periodic breathing in many patients is a temporary phenomenon. As outlined above, the two key parameters—chemoreflex gain and processing of signals in the brain stem—might be related to the influence of other brain centres.45,57

{kind=link}
{kind=link}
{kind=link}
{kind=link}
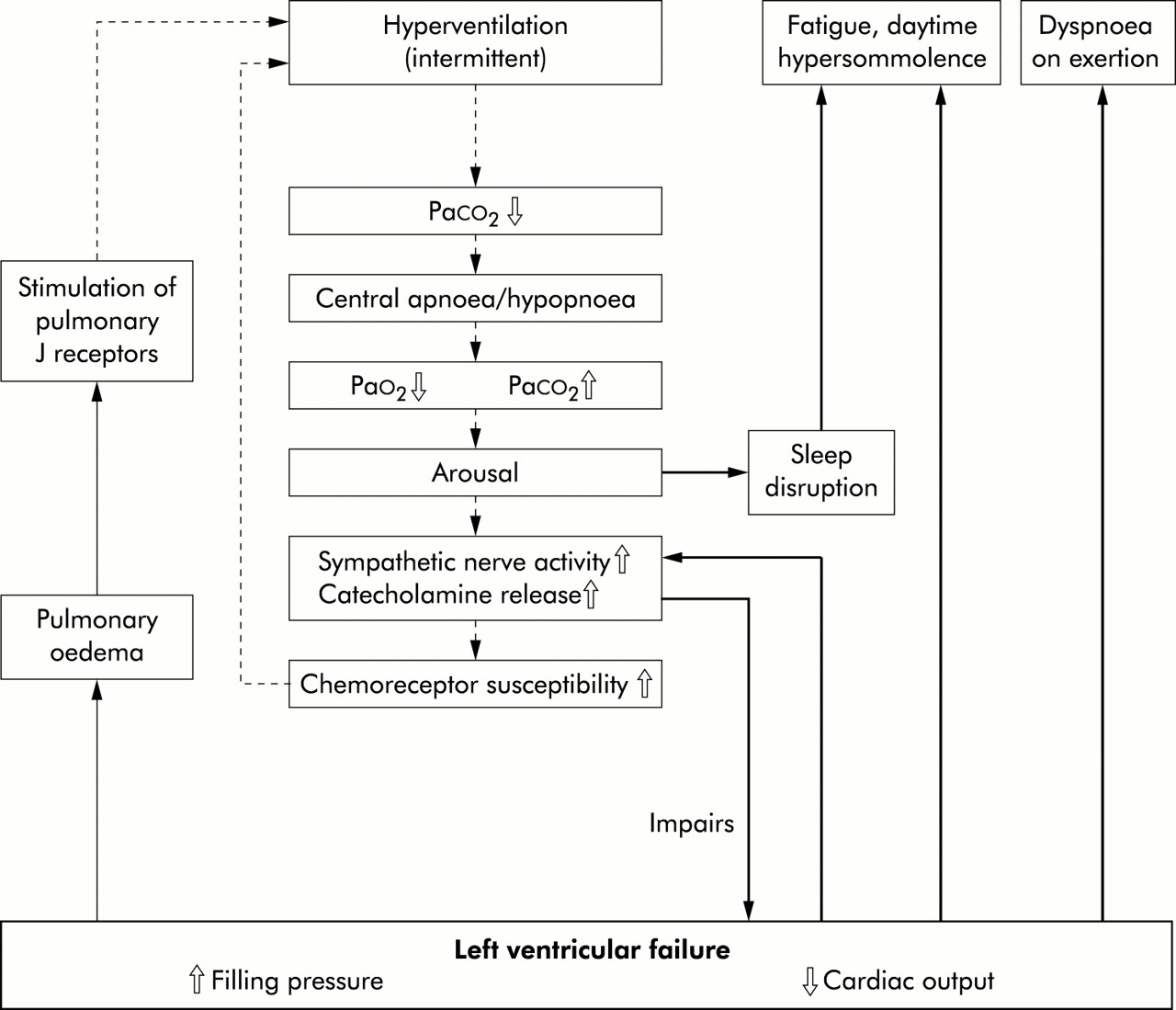
Interactions between left ventricular failure and instabilities in ventilation. The inner part of the flow chart shows the process during periodic breathing. Modified from Hall and Bradley.55
Haemodynamic factors presumably also play an important role in the pathogenesis of periodic breathing.58 During sleep the circulatory system is downregulated with a lower heart rate, arterial pressure, and cardiac output.59 When the patient is recumbent, an increase in preload may paradoxically reduce cardiac output by increasing functional mitral regurgitation as the mitral valve ring is stretched further. Alternatively, the attendant increased filling into the right ventricle and the redistribution and exacerbation of pulmonary oedema when lying supine may lead to increased right ventricular preload and afterload, respectively, thereby enhancing the reversed Bernheim effect which compromises left ventricular filling thus lowering cardiac output.60 The decreased cardiac output renders the patient susceptible to periodic breathing. Increasing cardiac output through vasodilator or positive inotropic therapy may reduce the incidence of periodic breathing.61
CONSEQUENCES OF PERIODIC BREATHING
During hypopnoea and apnoea the declining intra-alveolar oxygen content of the lung can cause an increase in right ventricular afterload (Euler-Liljestarand mechanism).62 The end systolic and end diastolic volumes of the right ventricle increase, producing a gradual shift of the interventricular septum towards the left ventricle (reversed Bernheim effect). This impairs ventricular filling and decreases the end diastolic volume of the left ventricle resulting in a diminished left ventricular output.63 It can be speculated that this is particularly likely if the myocardium is weakened by cardiomyopathy or ischaemic heart disease.
Arousals may also be associated with direct activation of the sympathetic nervous system.64 As a result, there is increased noradrenaline spillover in many organs and raised adrenaline and noradrenaline levels in the blood.65 The amount of catecholamines and their metabolites in overnight urine collections is proportional to the amount of periodic breathing and the number of arousals in patients with CHF and periodic breathing during sleep.66 Direct measurements of sympathetic activity in peripheral nerve fascicles revealed a close correlation between arousals in the cortical EEG and sympathetic nerve discharge.67 Further markers of sympathetic activation during arousal are a simultaneous increase in EEG activity, heart rate, and arterial blood pressure.4 Detailed analysis of heart rate variability around arousal reactions reveals cyclic sympathetic activation and vagal withdrawal.68–70 Chronically increased catecholamine concentrations resulting from structural impairment of the heart are therefore exacerbated by periodic breathing. This might precipitate cardiac arrhythmias and downregulate the expression of catecholamine receptors, and could result in a vicious circle with continuously increasing catecholamine levels.71,72 The unfavourable effects of endogenous catecholamines in CHF are widely recognised.73 Recent studies have shown that catecholamines induce a direct toxic effect on cardiac myocytes in vivo and subsequent myocardial fibrosis74 and an enhancement of cardiac myocyte hypertrophy.75
The most critical situation in periodic breathing is during late apnoea when arousal occurs. At this time the heart is supplied with the smallest amount of oxygen and sympathetic activation with rapid release of catecholamines exposes the heart to further stress. In patients with severe periodic breathing several hundred of these episodes may occur during one night. This has given rise to speculation that these repetitive increases in cardiac stress might contribute to the poorer prognosis of patients with periodic breathing.76
DIAGNOSIS
Full polysomnography is regarded as the “gold” standard for diagnosing CSAS. The typical ventilatory pattern of periodic breathing is identified visually according to the guidelines of the American Thoracic Society.3 It can usually be distinguished from the obstructive sleep apnoea syndrome which is characterised by cessation of oronasal air flow with persistence of thoracoabdominal ventilatory efforts. However, in some cases it can be difficult to exclude secondary airway collapse, which might occur in the late phase of a central apnoea. Patients with periodic breathing must be distinguished from those with mixed apnoeas of primarily obstructive aetiology. Obstructive apnoeas are sometimes followed by reflex central apnoeas, superficially mimicking primary central apnoea.77
Several indices describing the severity of the disorder have been established, such as the duration of periodic breathing as a proportion of the total sleep time, the apnoea-hypopnoea index (AHI) which calculates the mean number of events in 1 hour of sleep, the arousal index, and the oxygen saturation. Up to 15 apnoeic or hypopnoeic episodes per hour of sleep can be considered as normal or mild, 15–30 episodes per hour reflect moderate sleep apnoea, and an AHI of >30 indicates severe sleep apnoea.78
Screening systems which measure a limited number of sleep parameters and interpret the sleep study automatically without allowing direct visualisation of the raw data are not sufficient diagnostic tools for CSAS.79
TREATMENT
Before considering specific treatment for periodic breathing, medical treatment of the underlying heart disease must be optimised.80,81 Improvement in cardiac output has been shown to reduce periodic breathing.27,82 Other confounding conditions which might influence breathing during sleep (such as neurological disorders or drugs) must be ruled out.
Treatment for periodic breathing includes nocturnal non-invasive ventilation and oxygen application via nasal cannulae (table 1).83–90 Drugs such as theophylline, morphine derivatives, and supplementation of inhaled air with 3% carbon dioxide have been applied under experimental conditions in small studies.91–94 The prescription of sedatives for patients with any form of sleep disordered breathing should be discouraged. In these patients slow wave and REM sleep, which are the refreshing sleep stages, are already reduced due to frequent arousals. The use of benzodiazepines in particular can further reduce slow wave and REM sleep, resulting in worsening of daytime symptoms. By blunting arousal responses they may worsen nocturnal gas exchange.95
Key references for pathophysiology and treatment of central sleep apnoea syndrome (CSAS)
In the absence of major prospective randomised controlled trial data, the indications for treatment remain to be defined, but it should be considered when there are frequent and severe oxygen desaturations during sleep resulting from recurrent hypopnoeas or apnoeas, a high prevalence of arousals and sympathetic nervous system activation, and severe disruption of sleep architecture (reduced amount of slow wave or REM sleep) with subsequent daytime hypersomnolence.96
Oxygen
The application of oxygen at a rate of 4 l/min via nasal cannulae has been shown to influence nocturnal periodic breathing. Some investigators have reported an almost complete abolition of periodic breathing and an improved mean overnight oxygen saturation,89 whereas others observed only a partial (approximately 50%) reduction in the number of hypopnoeas and apnoeas.87,97 Andreas et al88 found a significant increase in exercise tolerance (peak oxygen consumption) in a cohort of 22 heart failure patients with periodic breathing during sleep after 1 week of nocturnal oxygen therapy. The same study revealed a significant correlation between the increase in peak oxygen consumption during ergospirometry and a decrease in CSR, in arousals, and in the amount of time spent in bed with an oxygen saturation <90%. Staniforth and colleagues98 conducted a randomised, double blind, crossover study comparing oxygen with room air in 11 patients with stable CHF. Four weeks of nocturnal oxygen treatment reduced the amount of apnoeas and periodic breathing during sleep, as well as the nocturnal urinary noradrenaline excretion. No significant changes in sleep quality, patient symptoms, or cognitive function were identified. It has been postulated that the limited effect of high oxygen concentrations is due to enhanced cellular metabolism in the peripheral chemoreceptors and a subsequent alteration of their setting.99
Continuous positive airway pressure (CPAP)
CPAP or non-invasive ventilation has been used for the treatment of patients with periodic breathing during sleep. Although the mechanism of action of CPAP is not fully understood in patients with CSAS, it has been shown to be an effective method of treatment.100 It can be difficult to introduce CPAP in some patients,101 but a relatively low pressure level of 3.7 mm Hg has been shown to be effective in adults with a normal body mass index.102 It stabilises the patient's ventilation and reduces the amount of periodic breathing, apnoeas, hypopnoeas, arousals, and the number of oxygen desaturations during sleep.84 The mean oxygen saturation increases and the transcutaneous carbon dioxide pressure increases back into the normal range.83
Studies of activation of the sympathetic nervous system during treatment with CPAP have shown a significant reduction in the excretion of noradrenaline, but not of adrenaline, in overnight urine collections.66 CPAP also has direct haemodynamic effects, increasing the intrathoracic pressure and thereby reducing cardiac afterload and preload by reducing venous return to the right atrium. This results in lower diastolic filling pressures and more effective pump function.103,104 CPAP recruits atelectatic lung areas, improving oxygen transfer in the lungs, and the higher intrapulmonary oxygen content might prevent recurrent rises of right ventricular afterload and subsequent pulmonary hypertension.105 However, a recently published randomised study investigating patients with CHF with and without periodic breathing revealed an improvement in left ventricular ejection fraction and in mortality in patients with periodic brething only, suggesting that any beneficial effect from CPAP is a consequence of an improvement in periodic breathing.106
In summary, current therapeutic approaches have focused on correction of the pathological breathing pattern. The effect of oxygen seems to be less successful than CPAP or non-invasive ventilation. There are no conclusive studies of the long term outcome of any treatment of CSAS. Many investigators prefer CPAP for treatment of periodic breathing because of the possible benefit of its direct mechanical effects on the failing heart.107 However, the development of new treatment strategies must be encouraged to find new approaches to the instability of the control of breathing in some patients with congestive cardiac failure.108 These may include different forms of non-invasive ventilation86 or drugs influencing the central control of carbon dioxide.47
CONCLUSIONS
CSAS with periodic breathing occurs frequently but is often unrecognised in patients with advanced CHF. In many patients, daytime fatigue and impaired physical performance is not just a symptom of the heart disease or side effect of treatment, but an indicator of sleep disordered breathing. The prognosis of patients with CHF seems to be significantly worse if CSAS is present. The causal relationship between CHF and CSAS is not fully understood, but delayed circulation time, increased chemoreceptor sensitivity to carbon dioxide, input into the respiratory centre from other brain centres and from peripheral receptors might have pathophysiological importance. Treatment options include CPAP and supplemental oxygen. CPAP is the most widespread treatment but, because of relatively poor patient acceptance in some studies, more sophisticated ventilatory support strategies need to be developed. Adequately powered prospective randomised controlled trials with survival and health status as end points are needed to establish whether the correction of sleep related abnormalities of breathing in patients with heart failure really does improve outcome.
Acknowledgments
TK was supported on a Long Term Research Fellowship funded by the European Respiratory Society. The authors thank Dr S B Pearson for helpful comments on the manuscript.