Article Text

Abstract
Background: Although inhaled corticosteroids have an established role in the treatment of asthma, studies have tended to concentrate on non-smokers and little is known about the possible effect of cigarette smoking on the efficacy of treatment with inhaled steroids in asthma. A study was undertaken to investigate the effect of active cigarette smoking on responses to treatment with inhaled corticosteroids in patients with mild asthma.
Methods: The effect of treatment with inhaled fluticasone propionate (1000 μg daily) or placebo for 3 weeks was studied in a double blind, prospective, randomised, placebo controlled study of 38 steroid naïve adult asthmatic patients (21 non-smokers). Efficacy was assessed using morning and evening peak expiratory flow (PEF) readings, spirometric parameters, bronchial hyperreactivity, and sputum eosinophil counts. Comparison was made between responses to treatment in non-smoking and smoking asthmatic patients.
Results: There was a significantly greater increase in mean morning PEF in non-smokers than in smokers following inhaled fluticasone (27 l/min v –5 l/min). Non-smokers had a statistically significant increase in mean morning PEF (27 l/min), mean forced expiratory volume in 1 second (0.17 l), and geometric mean PC20 (2.6 doubling doses), and a significant decrease in the proportion of sputum eosinophils (–1.75%) after fluticasone compared with placebo. No significant changes were observed in the smoking asthmatic patients for any of these parameters.
Conclusions: Active cigarette smoking impairs the efficacy of short term inhaled corticosteroid treatment in mild asthma. This finding has important implications for the management of patients with mild asthma who smoke.
- asthma
- smoking
- inhaled corticosteroids
Statistics from Altmetric.com
Inhaled corticosteroids are the most effective treatment currently available for chronic asthma.1 Patients with mild to severe asthma respond to inhaled corticosteroids as assessed by improved asthma symptoms and lung function as well as reduced bronchial hyperreactivity and eosinophilic inflammation.2–4 Both national and international asthma guidelines emphasise the importance of the early introduction of inhaled corticosteroids as first line treatment for those with mild disease.5, 6 A very small percentage of patients with asthma have been identified who do not benefit from corticosteroid treatment.7 The mechanisms of steroid resistance in these individuals are poorly understood but are thought to include abnormalities in glucocorticoid receptor function, corticosteroid pharmacokinetics, or transcription factor protein activity.7
Clinical studies in asthma have concentrated on non-smokers, but cigarette smoking is common with about 20% of asthmatics being regular smokers.8, 9 There is evidence for increased morbidity and mortality from asthma in individuals who are cigarette smokers.9–12 Asthmatic patients who smoke have been reported to have more severe asthma symptoms than non-smoking asthmatics,9, 11 an accelerated decline in lung function over time,10 increased hospital based care,13 and increased mortality following admission to hospital with an episode of near fatal asthma.12 There is comparatively little direct information about the effect of active smoking on drug treatment in asthma. In an uncontrolled study it was reported that improvements in airway function and plasma inflammatory markers in response to inhaled corticosteroid treatment might be attenuated in asthmatic smokers compared with asthmatic non-smokers.14
We have examined the effects of active cigarette smoking on the efficacy of inhaled corticosteroid treatment in subjects with mild asthma. To our knowledge, this is the first placebo controlled, randomised, prospective study of this issue. If steroid resistance is shown to be a feature of all asthmatic patients who actively smoke, this could have important implications for the treatment of these individuals.
METHODS
Patients and entry criteria
Twenty one non-smoking and 17 smoking asthmatic patients with no history of productive cough were recruited. Patients were treated only with inhaled bronchodilators as required, asthma was defined according to the American Thoracic Society definition,15 and bronchial hyperreactivity to methacholine was established with all subjects having a methacholine PC20 of less than 8 mg/ml. Bronchodilator reversibility to salbutamol was assessed and reversibility of >10% was a criterion for entry to the study if the forced expiratory volume in 1 second (FEV1) at baseline was <70% of predicted. No patient was recruited who had a history of upper respiratory tract infection or treatment with inhaled or oral corticosteroids in the 2 months prior to attendance. The study was approved by the West Ethics Committee, West Glasgow Hospitals University NHS Trust, and each subject gave written informed consent.
Study design
The randomised, double blind, crossover, placebo controlled study was of 10 weeks duration. Subjects attended the laboratory on five occasions, the first being a screening visit for consent, blood sampling (total serum IgE and serum cotinine levels), spirometric tests, and reversibility testing. Subjects kept morning and evening peak expiratory flow (PEF) recordings throughout the study, with the first week of the study being a placebo run in to establish baseline values. Thereafter there were three study periods, each of 3 weeks duration, during which subjects took inhaled placebo or fluticasone propionate (250 μg per puff) inhalers (metered dose inhalers via Volumatic spacer), two puffs twice daily according to randomisation. The patients were separated into smokers and non-smokers, with recruitment continuing in parallel for each group. At each study visit the PEF diary was retained, spirometric tests were performed, and a methacholine challenge test was carried out followed by sputum induction. The order of randomisation was such that, following a placebo run in of 1 week, each subject took either placebo or fluticasone propionate for 3 weeks followed by a placebo washout period of 3 weeks and a further 3 week period of placebo or fluticasone propionate. The order of treatment and placebo was balanced to minimise any order effect.
Measurements
Peak expiratory flow (PEF) recordings
PEF measurements were undertaken by patients at home using a mini-Wright peak flow meter (Clement Clarke, Harlow, UK). The best of three measurements was recorded twice daily (pretreatment) in the diary. Values of morning and evening PEF were averaged from the last 7 days before each study visit.
Spirometry
Forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) were measured with a dry spirometer (Vitalograph Ltd, Buckingham, UK) and the best of three attempts was taken for analysis. FEV1 was measured before and 20 minutes after 200 μg salbutamol to test reversibility on the first visit.
Bronchial hyperreactivity
Bronchial challenge testing was undertaken using the technique described by Cockcroft et al.16 In summary, methacholine was administered by nebulisation in doubling doses with measurement of spirometric parameters at each dose using a protocol and equipment standardised in our laboratory. Quantification of the response was calculated by linear interpolation and expressed as the provoking concentration of methacholine required to produce a 20% fall in FEV1 (PC20 methacholine).
Sputum induction
Sputum induction was performed using a modification of the method described by Pin et al.17 Briefly, after salbutamol 200 μg was administered by metered dose inhaler with large volume spacer, sputum induction was started using hypertonic (3%) saline administered via an ultrasonic nebuliser (Medix Ltd, UK) over a period of 20 minutes. The subjects were encouraged to expectorate at any time throughout the procedure and, in addition, inhalation was stopped every 5 minutes to allow expectoration and to allow spirometric tests to be carried out. The sample was collected in a sterile container and transferred to the laboratory on ice as soon as possible, and in all cases in less than 2 hours. The protocol dictates that, if FEV1 falls by more than 20%, the procedure is discontinued, although no subject in this study required discontinuation. All samples were processed without the laboratory staff being aware of the clinical information relating to the individual subject, and the procedure followed was similar to that described by Popov et al.18 Sputum samples were transferred to a Petri dish and the volume and macroscopic characteristics of the whole sample were recorded. Sputum plugs were selected to minimise salivary contamination,19 and treated with 4× volume of fresh 0.1% dithiothreitol (DTT) (Sputolysin; Calbiochem-Novabiochem (UK) Ltd, Nottingham, UK) in distilled water. Following incubation with DTT for 20 minutes the DTT treated samples were filtered through 50 μm mesh (R Cadoch & Sons, London, UK) to remove residual mucus clumps and a total cell count was made using a white cell counter (CBC5, Coulter Electronics Ltd, UK). An aliquot was removed, diluted to 106 cells/ml in phosphate buffered saline, and cytocentrifuged (500 rpm for 5 minutes) using a Shandon centrifuge. Differential cell counts were made from the resulting slides using Giemsa staining and expressed after exclusion of squamous epithelial cells which are taken to represent salivary contamination.19
Serum cotinine and total IgE
Ten ml venous blood was drawn and centrifuged prior to analysis of serum cotinine and total IgE. Serum cotinine was assayed by a commercially available enzyme linked immunosorbent assay (Cozart Biosciences, Abingdon, UK) and total IgE (international units/ml) by enzymatic immunoassay (Unicap System, Pharmacia, Uppsala, Sweden).
Assessment of compliance
Compliance was assessed by weighing inhalers on their return following each treatment period.
Statistical analysis
The intended power of the study was 80% (at the 5% level) to detect a mean treatment difference in PEF of 35 l/min with a standard deviation in non-smoking asthmatics of 40 l/min. Non-parametric statistics were used (Mann-Whitney U test) for comparisons of change in PEF and sputum eosinophil proportions, since these data were not normally distributed. The Student's t test was used to compare demographic and spirometric data. Bronchial hyperreactivity data were log transformed before analysis and are reported as geometric mean and geometric SD. Two types of analysis were performed: firstly, a comparison between smokers and non-smokers of changes across a particular treatment period (fluticasone propionate or placebo), assessing each treatment period therefore by measuring the change in values obtained immediately before and immediately after that particular treatment; and, secondly, a comparison across groups of the effect of fluticasone (fluticasone-placebo) in smokers and non-smokers. Significance was accepted at a level of p<0.05.
RESULTS
Baseline patient characteristics
Following screening for bronchial hyperreactivity and bronchodilator reversibility, 47 patients were enrolled into the study. Nine patients dropped out (all smokers) citing inconvenience in attendance as the main reason, leaving 38 patients for analysis (fig 1). There were no significant differences between smoking and non-smoking asthmatic patients at baseline in terms of age, duration of asthma, total serum IgE, FEV1 % predicted, bronchodilator reversibility, bronchial hyperreactivity (geometric mean PC20 methacholine), or baseline morning and evening PEF % predicted (table 1). Smoking asthmatics had higher mean (SD) serum cotinine levels than non-smokers (125.7 (13.1) mg/l v 11.4 (8.4) mg/l; p<0.0001).
Mean (SD) baseline patient characteristics
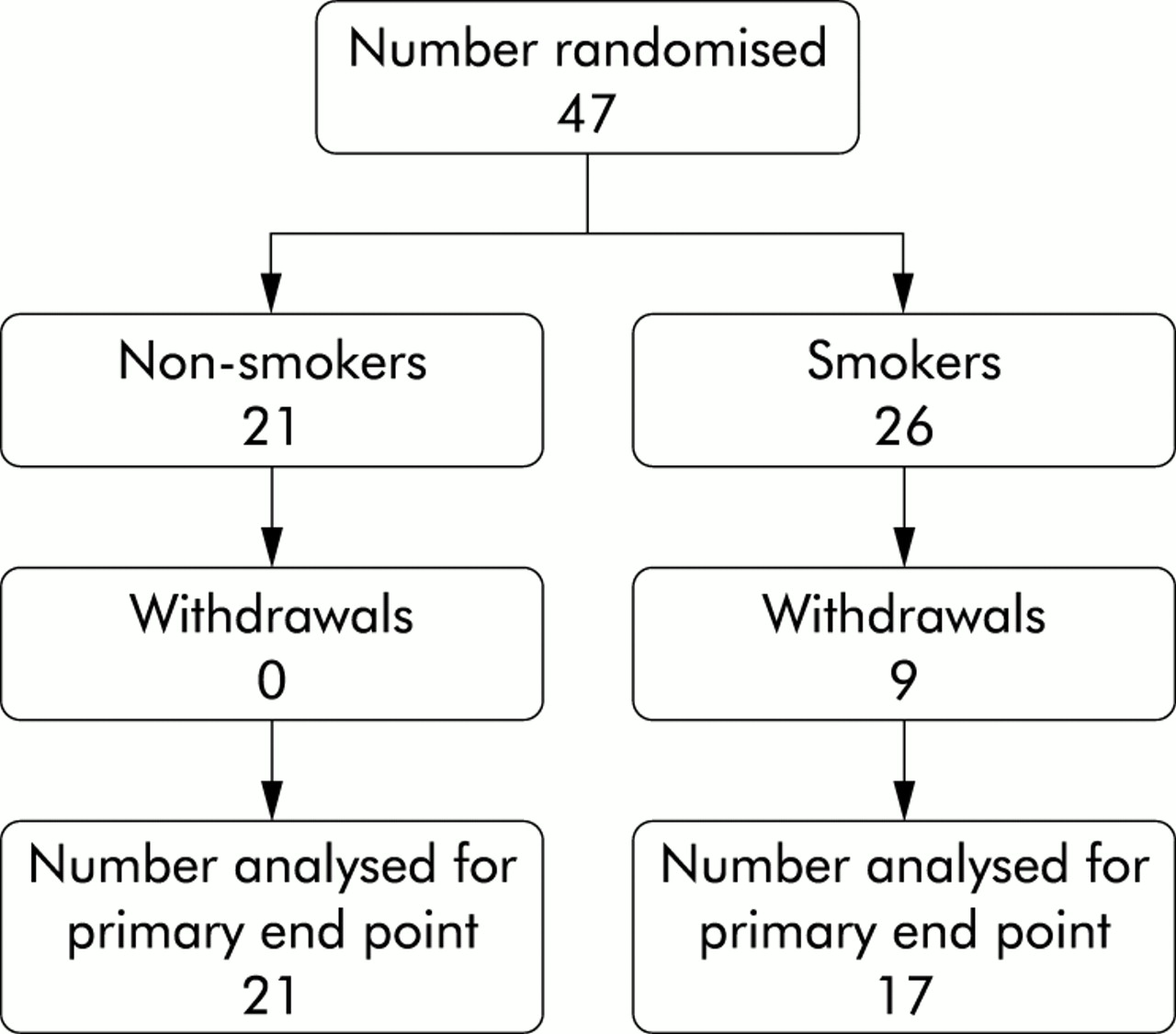
Design of trial.
Baseline measurements and comparison of change across treatment periods
PEF readings
There was no difference in baseline PEF measurements (% predicted) between non-smokers and smokers. In the non-smoking group the mean (95% CI) morning PEF increased from baseline by 27 (14.4 to 39.6) l/min following fluticasone and by 14 (–2.4 to 30.6) l/min following placebo (fig 2, table 2, p=0.016). In the smoking group the mean (95% CI) morning PEF decreased by –5 (–17.7 to 7.8) l/min following fluticasone and by 0 (–8.5 to 9.0) l/min following placebo (fig 2, table 2, p>0.05). There was a significantly greater change in morning PEF in non-smokers than in smokers following inhaled fluticasone (p=0.006). Evening PEF was not significantly affected by inhaled fluticasone in either group, although there was a trend towards a greater change in the non-smoking group than in the smoking group (p=0.051).
Mean (95% CI) changes in lung function and induced sputum eosinophil counts during each treatment period

{kind=link}
{kind=link}
Mean (95% CI) peak expiratory flow (l/min) in non-smoking and smoking asthmatic patients following treatment with inhaled placebo or fluticasone propionate 1000 μg per day. *p=0.016, greater than non-smokers after placebo; **p=0.001, greater than smokers after fluticasone.
Spirometry
In the non-smoking group the mean (95% CI) % predicted FEV1 increased from baseline by 5% (0.9 to 8.5) following fluticasone and by –1% (–4.3 to 2.2) following placebo (p=0.02, table 2), while in the smoking group the mean (95% CI) % predicted FEV1 decreased by –1% (–5.8 to 3.6) following fluticasone and by –1% (–5.7 to 4.7) following placebo (p>0.05). Changes of similar magnitude were observed for absolute values of FEV1, with a mean (95% CI) increase of 0.17 l (0.04 to 0.31) in non-smoking asthmatics following inhaled fluticasone and a mean (95% CI) decrease of –0.05 l (–0.18 to 0.07) following placebo (p=0.01). There was a trend towards a greater increase in FEV1 and FEV1 % predicted in the non-smoking group than in the smoking group in response to inhaled fluticasone (p=0.057).
Bronchial hyperreactivity
The non-smoking group had a geometric mean (SD) methacholine PC20 of 0.76 (1.3) mg/ml at baseline and a geometric mean (95% CI) increase in PC20 of 1.3 mg/ml (0.3 to 6.3) following inhaled fluticasone, compared with a geometric mean (95% CI) increase of –0.07 mg/ml (–0.7 to 0.9) following placebo (p=0.0002). The smoking group had a geometric mean (SD) methacholine PC20 of 1.5 (1.3) mg/ml at baseline and a mean (95% CI) increase in PC20 of –0.3 mg/ml (–1.5 to 6.1) following inhaled fluticasone compared with –0.3 mg/ml (–2.4 to 2.3) following placebo (no significant difference). Comparing non-smokers with smokers, there was no difference in the magnitude of change in methacholine PC20 following fluticasone.
Sputum eosinophils
The non-smoking group had a median (IQR) eosinophil proportion of 3.8 (0.7–5.5)% at baseline and a median (95% CI) decrease in the proportion of eosinophils of –1.8% (–6.6 to 2.5) following inhaled fluticasone and 0% (–0.7 to 2.5) following placebo (p=0.048). The smoking group had a median (IQR) eosinophil proportion of 0.3 (0–0.9)% at baseline (p=0.005 compared with non-smokers) and a median (95% CI) decrease in the proportion of sputum eosinophils of 0% (–0.5 to 1.4) following placebo and of 0% (–2.9 to 5.7) following inhaled fluticasone (no significant difference). The change in eosinophil counts following inhaled fluticasone was not significantly different in non-smoking and smoking asthmatic patients.
Comparison between end of placebo and fluticasone treatment periods
No significant changes in FEV1, PEF, methacholine PC20, or sputum eosinophils were found between the values at the end of the fluticasone and placebo treatment periods. We attribute this to the fact that the subjects had mild asthma and therefore the observed changes in response to an intervention were correspondingly small. A larger study would be required to show differences in this method of analysis.
Assessment of compliance
For all patients there was a reduction in inhaler weight for all study visits of >70% predicted.
DISCUSSION
We have demonstrated that smoking status has a significant effect on the response to inhaled corticosteroid treatment in patients with mild asthma. Non-smoking asthmatics experienced expected improvements in morning PEF, spirometric values, bronchial hyperreactivity, and sputum eosinophil proportions, whereas no significant changes in these parameters were found in the smoking asthmatic group.
The smoking subjects fulfilled the clinical criteria for the diagnosis of asthma,15 exhibited bronchial hyperreactivity, and had been symptomatic since their teens. Although cigarette smoking is the major identifiable factor in the development of COPD, we do not believe that the smoking asthmatic group in this study represents a group of subjects with COPD and a degree of reversibility. The difficulty of precisely differentiating subjects with asthma and mild COPD is accepted, but current guidelines suggest that an FEV1 of <80% is an appropriate diagnostic cut off for COPD while a normal FEV1 virtually excludes the diagnosis.20 The smoking asthmatics in this study had a mean baseline FEV1 higher than that typically associated with symptomatic COPD and, in addition, were younger, had bronchodilator reversibility, and a significant level of bronchial hyperreactivity to methacholine. Taken together, we would suggest that a diagnosis of asthma is entirely sustainable and was indeed the diagnosis made initially on the basis of symptoms and response to bronchodilator therapy.
The smoking and non-smoking patients were comparable in terms of demographic factors, morning and evening PEF (% predicted), bronchodilator reversibility, spirometric parameters, and serum total IgE, but there were some other distinguishing factors between the groups. The difference in the proportions of sputum eosinophils between smokers and non-smokers at baseline (median 3.8% v 0.3%) may indicate a difference in the nature of airway inflammation in smoking asthmatic patients compared with non-smokers, although we did not observe significantly different proportions of other sputum cell types in this study (data not shown). This could be a confounding factor for the differences observed in changes in sputum eosinophilia in response to inhaled corticosteroids between the groups. It is worth noting that neither group had high levels of sputum eosinophilia at baseline, and that the extent of the change in sputum eosinophils seen in the non-smoking asthmatics is small, although statistically significant, and should be interpreted appropriately. Bronchial biopsy studies would allow further investigation of the response of airway eosinophilia to treatment in similar subject groups while giving additional information about local changes in other inflammatory cells. This caveat does not, however, obviate the other findings of changes in morning PEF or bronchial hyperreactivity, and several studies have shown dissociation between the responses of airway function and airway inflammation to treatment with corticosteroids.21, 22 Sputum eosinophilia is a valuable means of monitoring the control of asthma23, 24; however, it is not a diagnostic test for asthma and sputum eosinophilia is not a universal finding even in exacerbations.25
Bronchial hyperreactivity was a criterion for entry into the study and, although not specific to the diagnosis of asthma, we believe that it is a useful indicator in patients who have no airflow obstruction or in those with only mild airflow obstruction.26, 27 Furthermore, the two groups could not be separated on the basis of factors which were used to establish the diagnosis of asthma. Smoke exposure was confirmed by the measurement of plasma cotinine levels, with all the non-smokers having values below the range generally accepted as indicative of active smoking and all the smokers having values within that range. Overall, we conclude that the baseline characteristics were matched between the two groups apart from smoking status, that the diagnosis of asthma as opposed to COPD is justifiable in the smoking group, and that active smoking is the most likely factor to account for the differences observed in response to treatment with inhaled corticosteroids.
The magnitude of the change in morning PEF in the non-smoking group in this study was comparable with other studies of the effect of inhaled steroids in asthma,28, 29 while the changes seen in FEV1 were slightly smaller than in some other studies.30 Although the reason for this difference is not known, the direction of change is as expected and is consistent with the observed change in bronchial reactivity in this study and in published data.31 Similarly, the reduction in the proportion of eosinophils is consistent with other studies of inhaled corticosteroids in mild asthma.28
Pedersen et al14 studied the responses to inhaled budesonide in asthma in a longer term study and found improvements in FEV1 and blood markers of inflammation which were not observed in a subgroup of smokers. The study was not placebo controlled, airway inflammation was not assessed directly, and the subgroup of asthmatic smokers studied had more severe airflow limitation than in the current study both before and after treatment. Our results are consistent with the findings of Pedersen's study, with additional direct information on airway inflammation and the additional validation of a placebo control. The asthmatic patients in our study had mild asthma and had relatively low cigarette exposure (mean 16.5 pack years), suggesting that the lack of effect of inhaled corticosteroids in smokers does not depend primarily on the severity of asthma or the extent of cigarette exposure. It seems unlikely that any lack of response in smokers could be attributed solely to an insufficient dose of inhaled corticosteroid, in view of the response of the non-smokers and the published evidence that fluticasone is effective at much lower doses than those used in this study.32 It has been shown that clinically significant effects of treatment are apparent after one day of treatment, and that the best observed effect occurred within 3 weeks of the start of treatment,29 suggesting that the time period allowed in this study should be sufficient to observe a clinical effect. Further studies, perhaps using oral corticosteroids, would be required to assess whether or not the attenuating effect of smoking could be overcome by increasing the dose of corticosteroid, increasing the duration of treatment, or both.
The mechanism behind the lack of response to inhaled corticosteroids in smoking asthmatics is not known. Cigarette smoke has the potential to cause harm to the airways in a number of ways, including direct toxicity and proinflammatory activity. Cigarette dose dependent inflammatory responses are observed in the airways of healthy adults33 and smokers exhibit altered airway cytokine regulation.34 The airway inflammation observed in smokers with COPD is characterised by a neutrophilia and is generally poorly responsive to inhaled or oral steroid treatment. In this group of mild asthmatics we did not observe a difference in the cellular profile of the sputum between smokers and non-smokers, but further investigation in more severe asthma or in heavier smokers might point towards an effect of this type. Other mechanisms by which cigarette smoking might prevent the anti-inflammatory actions of steroids include differences in drug access or clearance from the lungs due to increased mucus secretion or airway permeability. Alternatively, cigarette smoking might alter the molecular mode of action of steroids.
We conclude that cigarette smoking impairs responsiveness to inhaled corticosteroids in patients with mild asthma, and that studies assessing treatment in asthma should take smoking status into account in their conclusions about efficacy. The findings may have important clinical implications for asthmatic patients who smoke and further reinforce the need for smoking cessation in asthma, even in patients with mild disease.
Acknowledgments
The authors acknowledge the support of Chest Heart & Stroke Scotland which supported this work through a research fellowship, and Glaxo SmithKline (formerly Glaxo Wellcome) for the drug supplies.