Article Text

Abstract
Pulmonary alveolar proteinosis is a rare condition traditionally requiring treatment with whole lung lavage. The case is presented of a young man who obtained complete remission following treatment with granulocyte-macrophage colony stimulating factor, a new treatment option.
- pulmonary alveolar proteinosis
- treatment
- GM-CSF
Statistics from Altmetric.com
Case report
A 34 year old man presented in March 1998 with a 10 week history of malaise, cough productive of small quantities of clear sputum, and progressive shortness of breath on exertion such that he could no longer play touch rugby. He had no past medical history of note. His alcohol intake was significant at 30–40 units per week and he smoked 20 cigarettes per week.
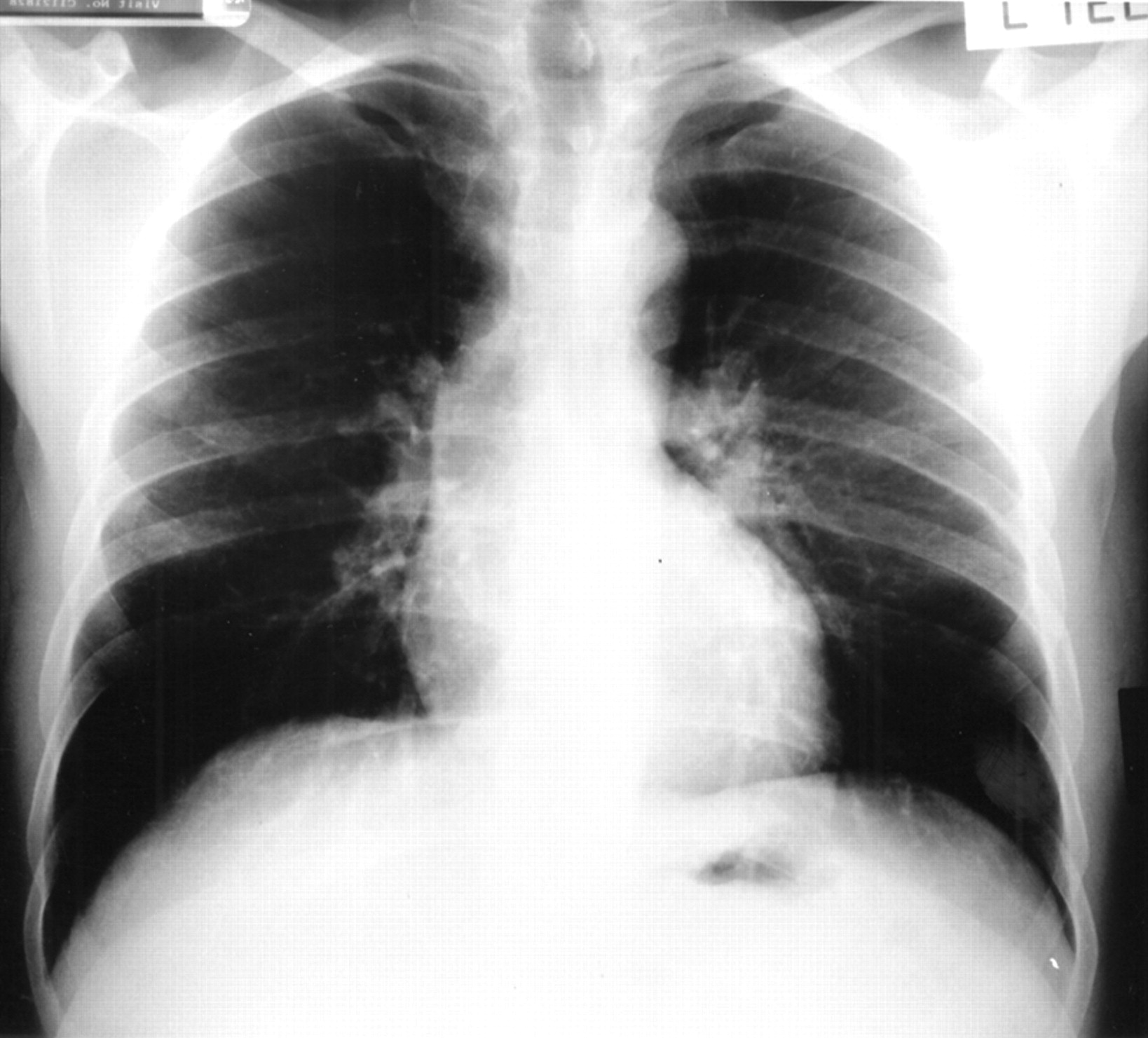
Physical examination and routine blood tests were normal. A chest radiograph showed bilateral pulmonary shadowing (fig 1). Spirometric tests showed a mild restrictive ventilatory defect with forced expiratory volume in one second (FEV1) 2.74 l (predicted 3.39 l) and forced vital capacity (FVC) 3.18 l (predicted 4.11 l). Oxygen saturation was normal. Static lung volume measurements revealed total lung capacity of 4.82 l (predicted 5.15 l) and residual volume of 1.54 l (predicted 1.36 l). Carbon monoxide transfer factor (Tlco) was reduced at 5.1 mmol/min/kPa (predicted 8.0) with transfer coefficient (Kco) relatively preserved at 1.31 mmol/min/kPa/l (predicted 1.52). At this stage a diagnosis of probable sarcoidosis was made.

Chest radiograph at presentation showing bilateral pulmonary infiltrates.
On review in early June 1998 his symptoms had shown little change. An exercise test demonstrated mild ventilatory restriction and limited exercise tolerance. His chest radiograph was unchanged, but a high resolution computed tomographic (CT) scan of the thorax showed bilateral pulmonary infiltrates and thickened interlobular septa (“crazy paving changes”; fig 2).

High resolution CT scan of the chest showing “crazy paving changes”.
Open lung biopsy performed at the end of June 1998 showed preservation of normal lung architecture with extensive deposition of granular eosinophilic material within alveolar spaces consistent with a diagnosis of pulmonary alveolar proteinosis. Electron microscopy was not performed. The condition and treatment options were considered and discussed with the patient who was strongly advised to give up smoking and was kept under regular review.
By March 1999 his symptoms had progressed such that he was unable to perform any useful work because of effort dyspnoea. Spirometric tests now showed FEV1 2.68 l and FVC 3.16 l. Po 2 on air was 76.4 mm Hg (10.2 kPa) and his chest radiograph showed an increase in the interstitial shadowing. He was commenced on daily treatment with granulocyte-macrophage colony stimulating factor (GM-CSF) by subcutaneous injection (5 μg/kg/day) in May 1999. This was well tolerated with only minor pain experienced at the injection site. His leucocyte count rose to a peak of 29.4 × 109/l (eosinophils 39%, neutrophils 52%) which improved following suspension of the treatment for 7 days. After 3 months of daily treatment with GM-CSF he was asymptomatic with normal spirometric values and arterial oxygenation, and resolution of the changes on the chest radiograph (fig 3), and has remained well with normal ventilatory function and a clear chest radiograph to date.
{kind=link}
{kind=link}
{kind=link}
Chest radiograph following 3 months of treatment with GM-CSF showing complete resolution of the pulmonary infiltrates.
Discussion
Pulmonary alveolar proteinosis (phospholipidosis) is a rare diffuse lung disease characterised by dense accumulation of large amounts of a phospholipoproteinaceous material in the alveoli with preservation of lung interstitium. It was first described in 1958 by Rosen et al 1 and is believed to involve defective surfactant homeostasis. Several different animal models exist, suggesting that pulmonary alveolar proteinosis is likely to represent a clinical syndrome rather than a single disease entity.2 ,3
The usual age at onset is in the third or fourth decade, and men are affected more than women in a ratio of 3:1. Cough and dyspnoea (usually on exertion) are the most common presenting symptoms, although a low grade fever in the absence of infection may also be noted. Physical examination is often normal, but may reveal clubbing, cyanosis, or inspiratory crackles.4 Diagnosis has traditionally relied on open lung biopsy.5
Although up to 25% of cases may show evidence of spontaneous remission, treatment is generally accepted to be indicated if a subject experiences limitations in daily activities due to dyspnoea. Whole lung lavage is currently accepted as the most effective form of treatment for advanced cases.6 Corticosteroids, potassium iodide, and streptokinase have all been tried with variable results.
Although prognosis with whole lung lavage is generally good, the treatment is both time consuming and technically demanding. There are no randomised controlled clinical trials reported in the literature, but response rates are typically around 60%—that is, 60% of patients subjectively return to normal in terms of exertional capacity after two washes per lung. A small proportion (<15%) require lavage every 6 months and fewer than 10% do not respond.6
Previous reports have suggested a potential role for GM-CSF in the treatment of pulmonary alveolar proteinosis.7-10 This is based on the observation that mice deficient in the gene for GM-CSF develop alveolar accumulations of surfactant substances similar to that seen in pulmonary alveolar proteinosis.2 The exact mechanism is unknown but may be due to the absence of macrophage activation by locally synthesised GM-CSF. To date, six cases have been reported in the literature with partial response to this novel treatment.
Our patient showed progressive deterioration over the course of several months until he was severely limited by dyspnoea. This trend was only reversed after starting treatment with GM-CSF. The treatment was well tolerated and complete clinical remission was achieved after 3 months.
We believe that this was not a case of spontaneous remission and suggest that this provides further evidence that GM-CSF may be an effective alternative to whole lung lavage in selected cases of pulmonary alveolar proteinosis. Furthermore, we believe that this is the first documented case of complete remission induced by GM-CSF in this rare condition.