Article Text

Statistics from Altmetric.com
Pulmonary lymphoid lesions encompass a spectrum of inflammatory and reactive lesions that are often difficult to diagnose since they are difficult to differentiate from other reactive and neoplastic entities (box 1). Understanding these lymphoid lesions is complicated by the evolution of concepts, criteria, and terminology over the past few decades. This review will attempt to summarise a historical perspective on this subject and present the current concepts of pulmonary lymphoid lesions.
Non-neoplastic
Intrapulmonary lymph node
Follicular bronchiolitis (diffuse lymphoid or MALT hyperplasia)
Lymphocytic interstitial pneumonia
Nodular lymphoid hyperplasia
Castleman's disease
Neoplastic
malignant lymphoma
Non-Hodgkin's lymphoma
- (1)
- B cell
Extranodal marginal zone B cell lymphoma of MALT type
Lymphomatoid granulomatosis
Diffuse large cell lymphoma
Lymphoplasmacytic lymphoma
Plasmacytoma
- (2)
- T cell
Peripheral T cell lymphoma
Anaplastic large cell lymphoma
- (3)
- Special types of lymphoma
Intravascular lymphoma
Primary effusion lymphoma
Hodgkin's lymphoma
leukaemic infiltration
Lymphocytic leukaemia, acute and chronic
Non-lymphocytic leukaemia, acute and chronic
Granulocytic sarcoma
Box 1 Lymphoid lesions of the lung. MALT = mucosa associated lymphoid tissue.
This review will focus on the major pulmonary non-neoplastic lymphoid lesions which include intrapulmonary lymph nodes, follicular bronchitis/bronchiolitis, lymphocytic interstitial pneumonia, and nodular lymphoid hyperplasia.1-3 The terms “lymphocytic interstitial pneumonia” or “diffuse lymphoid hyperplasia” have sometimes been used for both lymphocytic interstitial pneumonia and follicular bronchiolitis. However, in this review lymphocytic interstitial pneumonia and follicular bronchiolitis are regarded separately.
Intrapulmonary lymph nodes
Intrapulmonary lymph nodes in the periphery of the lung are usually situated in a subpleural location or adjacent to interlobular septa.2 In one necropsy study they were found in the lungs of 18% of patients.4
CLINICAL AND RADIOLOGICAL FEATURES
They are usually discovered as an incidental radiographic finding in an asymptomatic patient.2 Most patients who undergo resection for intraparenchymal lymph nodes are middle aged or older and have a history of cigarette smoking and organic dust exposure.2 They may be solitary or multiple.
PATHOLOGICAL FEATURES
Grossly, they appear as grey, brown, or black round to oval-shaped nodules beneath the pleura. Histologically, they consist of circumscribed nodules of lymphoid tissue that are not encapsulated, but usually border on the pleura or interlobular septa (fig1).2 Germinal centres may be seen but are frequently absent (fig 1). Histiocytes containing dust pigment and/or birefringent particles are usually present and are often prominent. The birefringent particles typically have a needle-like shape and represent silica. According to Kradin and Mark,2 50% of surgically resected intrapulmonary lymph nodes contain hyalinised silicotic nodules and ectatic lymphatic channels. Based on this observation, they speculated that development of intrapulmonary lymph nodes results from the presence of inorganic dust and lymphatic obstruction.

Intrapulmonary lymph nodes. (A) Intrapulmonary lymph node consisting of a circumscribed, nodular, subpleural mass of lymphoid tissue with hyperplastic lymphoid follicles. (B) Abundant anthracotic pigment is present between the aggregates of lymphoid tissue.
Follicular bronchiolitis
Follicular bronchiolitis is also known as hyperplasia of the bronchial associated lymphoid tissue (BALT) or hyperplasia of the mucosal associated lymphoid tissue (MALT), and pulmonary lymphoid hyperplasia. Bienenstock et al have published several classical studies of the BALT.5 ,6 They described the lymphoid aggregates along the bifurcation of the bronchioles and along the lymphatic routes. The lymphoepithelium was defined as the mucosal epithelium, which often contained infiltrating lymphocytes and showed attenuation and flattening. Follicular bronchiolitis results from antigenic stimulation of the BALT and polyclonal lymphoid hyperplasia.5-7
CLINICAL AND RADIOLOGICAL FEATURES
Follicular bronchiolitis occurs in several clinical settings including collagen vascular disease, immunodeficiency, hypersensitivity disorders, and in a variety of non-specific airway centred inflammatory conditions such as bronchiectasis, airway obstruction, and infections.8 ,9 It can occur in any of the collagen vascular disorders but is especially seen in rheumatoid arthritis.10-15 BALT hyperplasia has also been reported in immunodeficiency syndromes such as AIDS,8 ,16 IgA deficiency, Evan's syndrome (autoimmune haemolytic anaemia and immune thrombocytopenia),17 and common variable immunodeficiency syndrome.9 A familial example of follicular bronchiolitis has been reported under the term “familial pulmonary nodular lymphoid hyperplasia”.18
Follicular bronchiolitis occurs mostly in adults8 ,9 but can also be found in children.9 ,19 Yousemet al reported a mean age of 44 years (range 6–69) for patients with collagen vascular disease, 16 years (range 1.5–32) for patients with immunodeficiency syndromes, and 55 years (range 38–77) for patients with hypersensitivity syndromes.9 The typical presenting symptom is progressive shortness of breath and cough, but some patients may have fever, recurrent pneumonia, or weight loss.8 ,9 Pulmonary function may show obstruction, restriction, a mixed pattern, or no defect.9 Patients with hypersensitivity syndromes frequently have a peripheral blood eosinophilia.9
Chest radiographs show bilateral reticular or reticulonodular infiltrates.9 One study of high resolution CT scans in 12 patients with follicular bronchiolitis showed bilateral centrilobular nodules in all patients and peribronchial nodules in almost half of the cases.8 Nodules smaller than 3 mm in diameter were present in all patients, but half the patients also had larger nodules of 3–12 mm diameter. Ground glass opacities were seen in 75% of patients.8
Patients with follicular bronchiolitis typically have a favourable prognosis. However, Yousem et al 9 found that patients under 30 years of age tended to have progressive disease. Treatment may be directed at the underlying disease or may consist of steroids or azathioprine. Death associated with disease occurred in only two of 19 patients reported by Yousem et al,9 both of whom had collagen vascular disease and developed bronchopneumonia.
PATHOLOGICAL FEATURES
Histologically, follicular bronchiolitis consists of abundant peribronchiolar lymphoid follicles (fig 2).9 The lymphoid follicles may be situated between bronchioles and pulmonary arteries causing compression of the airway lumen, and may also be present along the interlobular septa and beneath the pleura. The epithelium of the bronchioles is often infiltrated by lymphocytes. The lymphoid infiltrate is limited to the peribronchiolar lymphoid follicle or it may extend superficially into the surrounding alveolar interstitium, but it lacks the extensive alveolar septal infiltration of lymphocytic interstitial pneumonia. However, there is a continuum of lymphoid infiltration between follicular bronchiolitis and lymphocytic interstitial pneumonia and in some cases this distinction may be somewhat arbitrary. A neutrophilic exudate is present within the bronchiolar lumen in approximately 50% of cases. Secondary obstructive changes such as foamy macrophages and small foci of organising pneumonia may be seen.9 Follicular bronchiolitis is frequently a secondary lesion and may be associated with other findings such as bronchiectasis, bronchiolectasis, bronchiolar fibrosis. or organising pneumonia.

Follicular bronchiolitis. (A) Hyperplastic lymphoid tissue surrounding the bronchiole. Mild bronchiolar fibrosis is also present. (B) Hyperplastic lymphoid tissue appears to compress the lumen of one bronchiole (centre right). The adjacent bronchioles show marked chronic inflammation. There is mild bronchiolectasis and chronic inflammation of a bronchiole in the surrounding lung parenchyma.
Sato et al 14 characterised the cellular distribution of lymphoid cells in follicular bronchiolitis in patients with rheumatoid arthritis. They distinguished four distinct regions of the BALT including the lymphoepithelium, the dome area, the follicular area, and the parafollicular area. The lymphoepithelial and dome areas of the BALT are associated with the bronchiolar mucosa. In the follicular area they found mostly B cells expressing surface IgM. However, in the dome and parafollicular areas they found B cells expressing IgA. The parafollicular area consisted mostly of T cells, more CD4+ than CD8+, most of which expressed the T cell αβ receptor antigen. Based on this finding, which is similar to results in diffuse panbronchiolitis, the authors suggested that follicular bronchiolitis in rheumatoid arthritis is related to extrinsic stimulation as well as alterations of the immune response.14
Lymphocytic interstitial pneumonia
Lymphocytic interstitial pneumonia is a form of interstitial pneumonia that is characterised by diffuse infiltration of the alveolar septa by a dense lymphocytic infiltrate. Several terms have been used for this lesion including “lymphoid interstitial pneumonia” and “plasma cell interstitial pneumonitis”. In 1969 Liebow described lymphocytic interstitial pneumonia as a form of interstitial pneumonia to be separated from other interstitial pneumonias such as usual interstitial pneumonia and desquamative interstitial pneumonia.20 However, a substantial percentage of the lesions that were classified by Liebow as lymphocytic interstitial pneumonia21 were subsequently recognised to be low grade B cell lymphomas and today they would be classified as MALT lymphomas.22 ,23 As a result, lymphocytic interstitial pneumonia was excluded from the classifications of idiopathic interstitial pneumonias for several decades. However, lymphocytic interstitial pneumonia exists as an inflammatory and non-neoplastic process and it may rarely be idiopathic. It has therefore been included in an ATS/ERS international multidisciplinary consensus classification of idiopathic interstitial pneumonias.24
CLINICAL AND RADIOLOGICAL FEATURES
Lymphocytic interstitial pneumonia is associated with a variety of conditions including dysproteinaemia, autoimmune disorders, collagen vascular diseases, bone marrow transplantation, and the acquired immunodeficiency syndrome, but true idiopathic lymphocytic interstitial pneumonia is very rare (box 2).
Collagen vascular disease
Other immunological disorders
Pernicious anaemia58
Myasthenia gravis21
Hashimoto's thyroiditis33
Primary biliary cirrhosis23
Coeliac sprue59
Immunodeficiency
Infections
Drug induced/toxic exposure
Dilantin (phenytoin)68
Box 2 Clinical conditions associated with the lymphocytic interstitial pneumonia pattern.
In patients with associated conditions the underlying disease usually dominates the clinical presentation of lymphocytic interstitial pneumonia. The presentation of idiopathic lymphocytic interstitial pneumonia is not well defined.24 It occurs most often in the fourth to sixth decade of life21 ,25 ,26 and women are affected more than men. An example of familial lymphocytic interstitial pneumonia has been reported in two brothers who presented during childhood.27 Lymphocytic interstitial pneumonia is rare in HIV infected adults, but in children under the age of 13 it is one of the defining criteria for AIDS proposed by the Center for Disease Control.28
The most common symptoms are gradual onset of cough and dyspnoea. Other possible manifestations include weight loss, fever, chest pain, and arthralgias. Lymphadenopathy is found mostly in patients with Sjögren's syndrome.25 Pulmonary function often reveals a restrictive ventilatory defect with a low transfer factor. If laboratory analysis reveals a monoclonal gammopathy or hypogammaglobulinaemia, the index of suspicion for a lymphoproliferative malignancy should be raised.
Little is known about the clinical behaviour of idiopathic lymphocytic interstitial pneumonia. If there is an underlying disease, this will determine the clinical course. Optimal treatment is administration of corticosteroids, which usually results in improvement or resolution of symptoms.25 ,29 Several complications of lymphocytic interstitial pneumonia have been reported in HIV infected patients with lymphocytic interstitial pneumonia, including recurrent pneumococcal pneumonia,30 bronchiectasis,31 and lung cyst formation.32
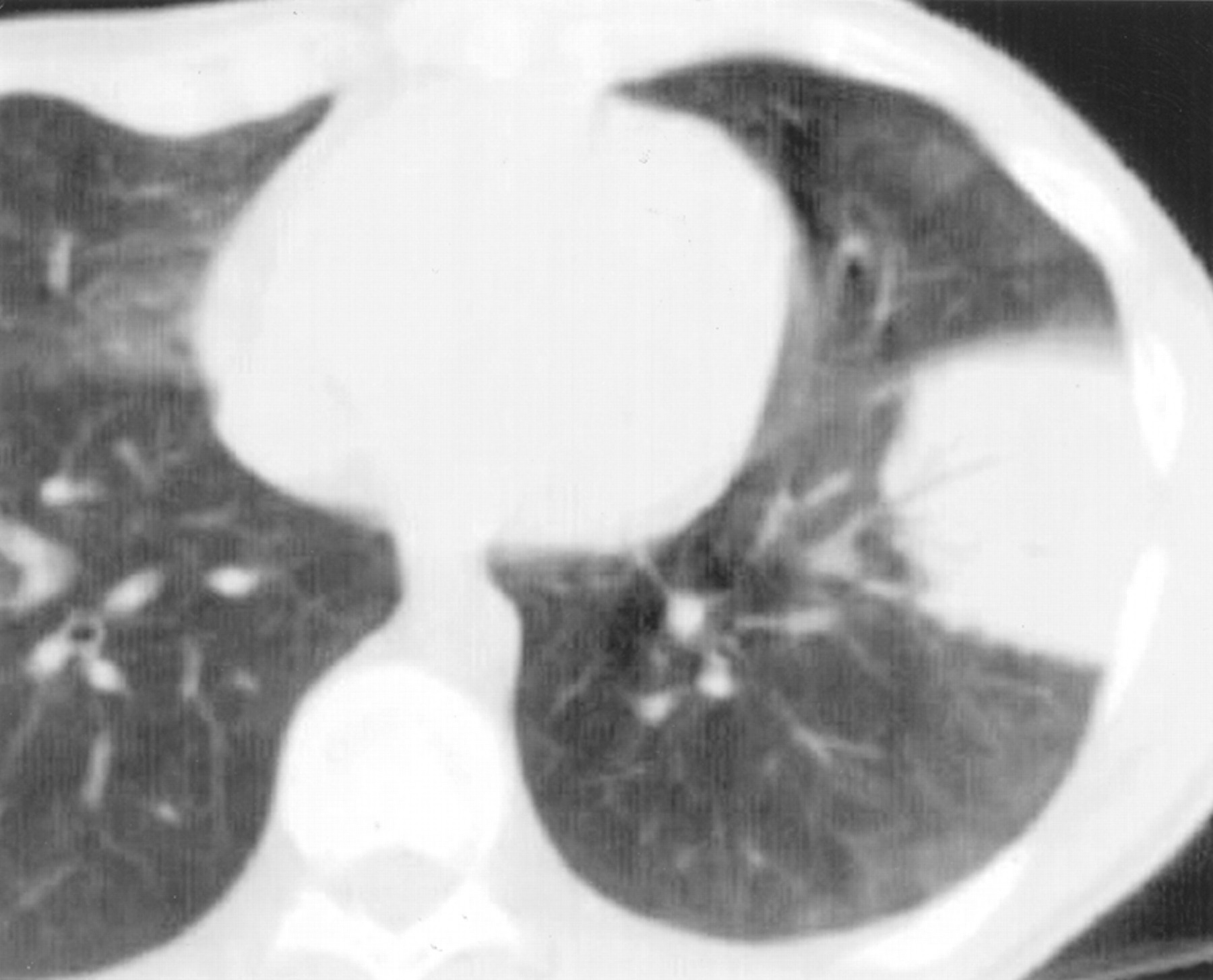
Chest radiographs may show basilar infiltrates with an alveolar component or diffuse infiltrates associated with honeycombing.33 High resolution CT scans typically show ground glass opacities (fig 3). Nodular lesions may be seen and, rarely, cysts may occur in a peribronchovascular location (fig3).32
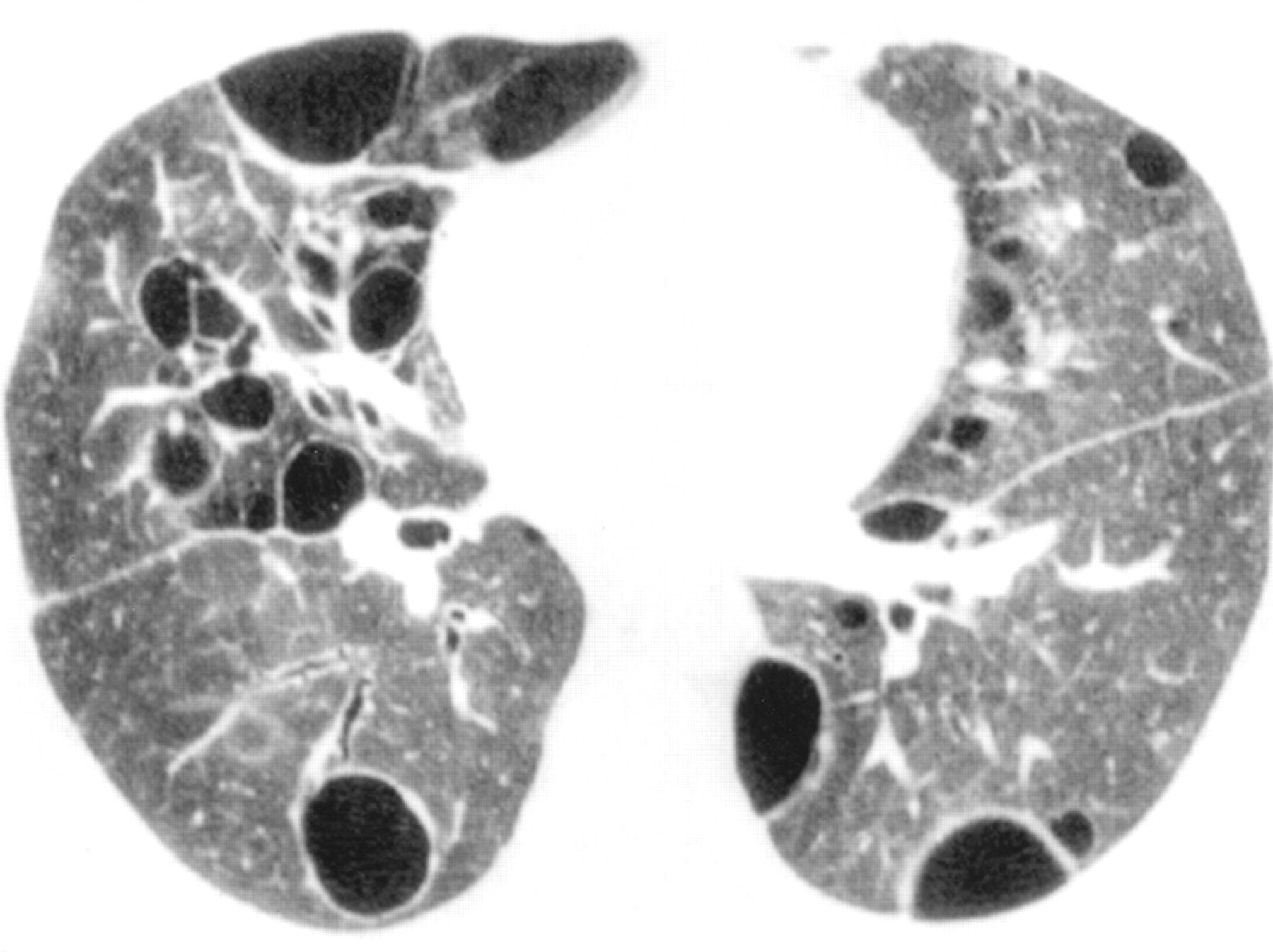
Lymphocytic interstitial pneumonia. An axial CT section through the lower lobes showing diffuse hazy ground glass opacity and multiple thin walled cysts.
PATHOLOGICAL FEATURES
Lymphocytic interstitial pneumonia is characterised by a marked lymphoid infiltrate with extensive involvement of the alveolar septa (box 3, fig 4). The lymphoid infiltrate consists mostly of lymphocytes with varying numbers of plasma cells. The process typically involves lung biopsy specimens diffusely. In some cases there may be sparing of some lung parenchyma if the lesions are somewhat nodular, but the involved areas show diffuse alveolar septal infiltration. Type II pneumocyte hyperplasia may be present. Lymphoid follicles, including follicles with germinal centres, are often present, usually in the distribution of pulmonary lymphatic vessels or bronchioles (follicular bronchiolitis). The lymphoid cells consist of both B and T cells. The T cells occur mostly in the alveolar septal interstitium while the B cells are mostly found in the lymphoid follicles.23 ,34Interstitial fibrosis and small foci of organising pneumonia may be present. Non-necrotising granulomas may also be seen. Epstein-Barr virus has been identified in lung biopsy specimens from both HIV infected and non-infected patients with lymphocytic interstitial pneumonia.35-37
Major features
Diffuse interstitial infiltration of involved areas
Predominantly alveolar septal distribution
Infiltrates comprising mostly T lymphocytes, plasma cells, and macrophages
Lymphoid hyperplasia (BALT hyperplasia) frequent
Pertinent negative features
Lack of tracking along lymphatic routes (bronchovascular bundles, pleura and interlobular septa) characteristic of lymphomas
Organising pneumonia inconspicuous or absent
Lack of Dutcher bodies (intranuclear inclusions in B lymphocytes)
Lack of monoclonal light chain staining pattern of plasma cells (polyclonal pattern present)
Lack of extensive pleural involvement or lymph node involvement
Lack of necrotising granulomas
Box 3 Histological features of lymphocytic interstitial pneumonia.24
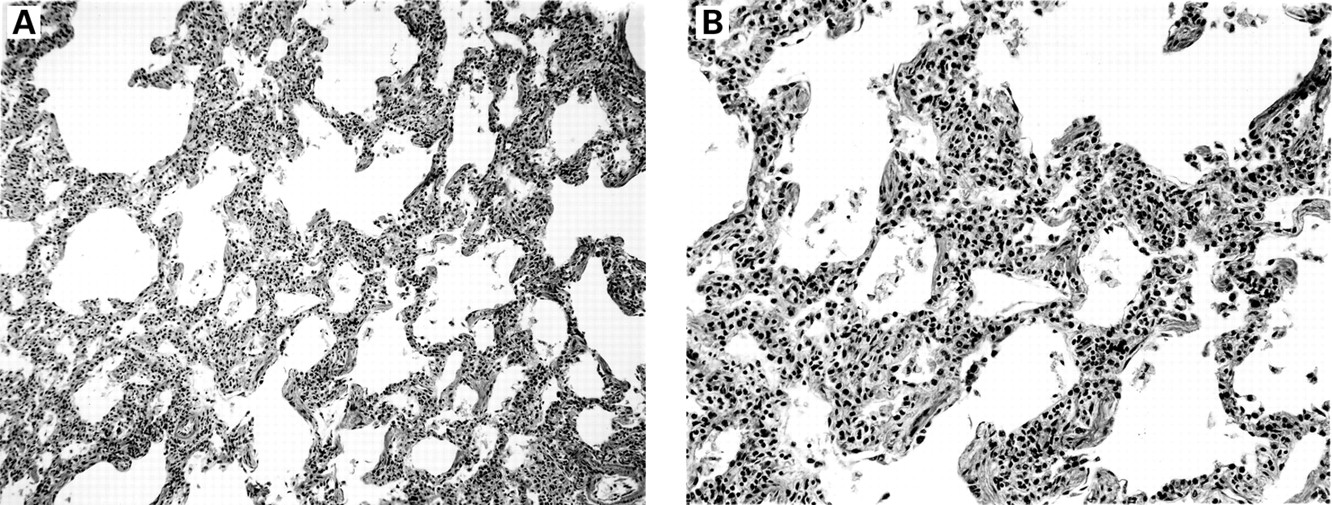
Lymphocytic interstitial pneumonia. (A) The lung is diffusely infiltrated by lymphocytes and plasma cells. (B) The inflammatory cells infiltrate the alveolar septa diffusely.
Lymphocytic interstitial pneumonia must be differentiated histologically from malignant lymphoma, follicular bronchiolitis, nodular lymphoid hyperplasia, and infection. It must also be distinguished from other interstitial lung disorders such as non-specific interstitial pneumonia, organising pneumonia, and usual interstitial pneumonia. The term “lymphocytic interstitial pneumonia” has been used previously for cases of follicular bronchiolitis with extensive lung involvement, even in the absence of infiltration of the alveolar septa, but this does not fit our current definition.2 There is a continuum of lymphoid infiltration between follicular bronchiolitis and lymphocytic interstitial pneumonia, but in most cases the patterns can be distinguished34 and the term “lymphocytic interstitial pneumonia” is limited to those cases with extensive alveolar septal infiltration.24
The major differential diagnosis from a clinical standpoint is the separation of lymphocytic interstitial pneumonia from low grade lymphoma. This distinction can be difficult on haematoxylin and eosin stained sections and may require immunohistochemical analysis and molecular gene rearrangement studies such as polymerase chain reaction testing for clonal rearrangement of the immunoglobulin heavy chain gene.34 ,38 The malignant lymphomas that are likely to be confused with lymphocytic interstitial pneumonia are extranodal marginal zone B cell lymphoma of MALT and small lymphocytic lymphoma. In contrast to lymphocytic interstitial pneumonia, the lymphoid infiltrate in malignant lymphoma is more dense and monomorphous and it may show destruction of lung architecture. Other features that favour lymphoma include the presence of tracking along lymphoid routes, infiltration of the parietal pleura and lymph nodes, and the presence of Dutcher bodies (intranuclear inclusions in B lymphocytes). A complicating feature of low grade lymphomas is that there are frequently reactive lymphoid follicles and lymphoid infiltrates at the periphery that may give a misleading impression of a reactive process. Rarely does lymphocytic interstitial pneumonia progress to malignant lymphoma. In most cases where this has been reported, the lesion may have represented malignant lymphoma from the outset.39 ,40
Certain infections can cause a histological pattern of lymphocytic interstitial pneumonia, especially Pneumocystis carinii pneumonia.41 For this reason, special stains should be performed on cases with marked lymphoid infiltrates to exclude the presence of microorganisms.
The lymphocytic interstitial pneumonia pattern must also be separated from the patterns of hypersensitivity pneumonitis, cellular non-specific interstitial pneumonia, and organising pneumonia. The pattern in hypersensitivity pneumonitis differs from lymphocytic interstitial pneumonia in that the lymphoid infiltrates are less prominent, there is a peribronchiolar distribution, and poorly formed granulomas and organising intraluminal fibrosis are often present.22 The cellular pattern of non-specific interstitial pneumonia consists of a mild to moderate interstitial lymphocytic and/or plasma cell infiltrate,42 but it falls short of the extensive alveolar septal infiltration seen in lymphocytic interstitial pneumonia. Before the description of the cellular pattern of non-specific interstitial pneumonia,42 ,43 some of these cases were probably included in reports of lymphocytic interstitial pneumonia. Based on the narrower definition of lymphocytic interstitial pneumonia, the existing literature on this condition may need to be reassessed.
Nodular lymphoid hyperplasia
The existence of a benign nodular lymphoid proliferation in the lung has been a controversial subject. Since the 1960s most nodular lymphoid proliferations were regarded as benign. Salzstein described pulmonary pseudolymphoma in 1963 as a nodular lymphoid proliferation that had numerous germinal centres and a benign clinical course.44 However, as with lymphocytic interstitial pneumonia, it became recognised in the 1980s that many of these cases were actually MALT lymphomas45-47 and some doubted whether a benign nodular lymphoid lesion even existed.45However, Kradin and Mark described such a lesion under the term “nodular lymphoid hyperplasia” in 1983,2 and the 1999 Histologic Typing of Lung and Pleural Tumors by the World Health Organization and International Association for the Study of Lung Cancer recognised this as a pulmonary entity to replace the former term of “pseudolymphoma”.48 The existence of nodular lymphoid hyperplasia was validated in a detailed study by Abbondanzoet al in 2000.1
CLINICAL AND RADIOLOGICAL FEATURES
Abbondanzo et al reported 14 cases with a slight female predominance (4:3) and a median age of 65 years (range 19–80).1 The most common presentation in 71% of cases was as an incidental chest radiographic finding. In 64% of cases there was a solitary lesion (fig 5) but in 36% of cases the lesions were multiple.1 Multiple bilateral lesions are uncommon. When symptomatic, patients have shortness of breath, cough, and/or pleuritic chest pain.

Nodular lymphoid hyperplasia. An axial CT section at the level of the left ventricle showing a well circumscribed pleural based mass with typical air bronchograms.
Surgical resection is curative for nodular lymphoid hyperplasia.1 Nevertheless, there may be cases in which it is difficult to entirely exclude a diagnosis of MALT lymphoma and the patients should be followed carefully. This is particularly true for patients who have multiple bilateral lesions showing growth. Even patients with MALT lymphomas have an excellent prognosis, so the follow up period may need to extend for many years.
PATHOLOGICAL FEATURES
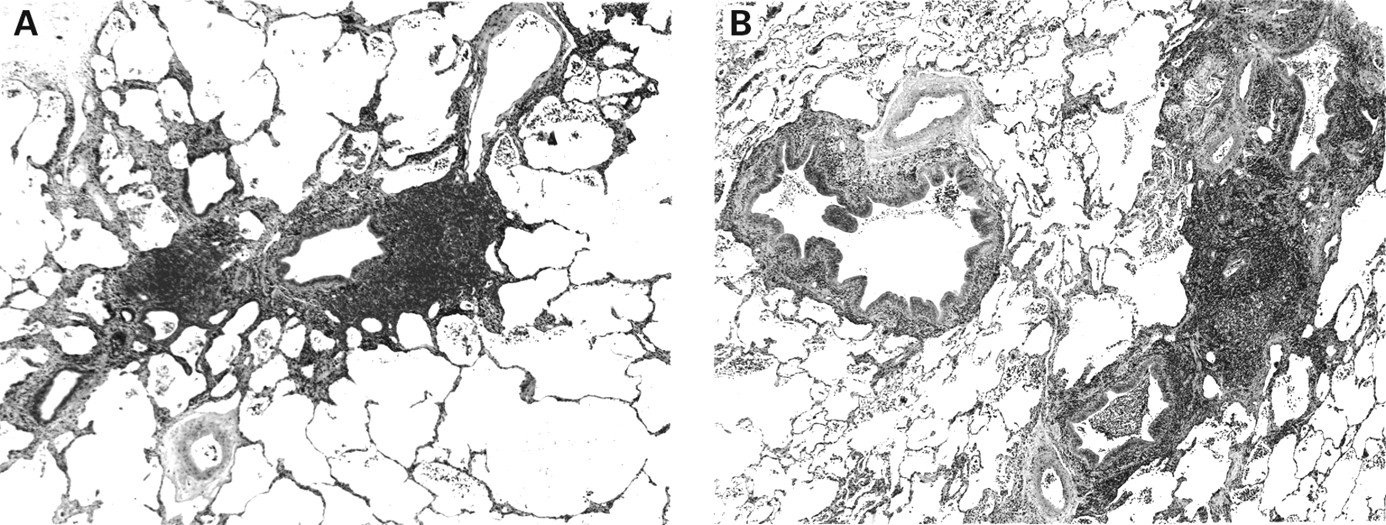
Nodular lymphoid hyperplasia is most often situated in a subpleural location.1 It consists of well demarcated nodules of grey, white, or tan tissue which on palpation feel firm, rubbery, or fleshy. Most cases are solitary, but two or three nodules may be present. The mean size measures 2.1 cm (range 0.6–6).1 Histologically, the lesions are circumscribed masses of lymphoid tissue with numerous reactive germinal centres (fig6). The germinal centres typically have well preserved mantle zones and plasma cells are prominent within the interfollicular areas (fig 6B and C). Russell bodies (large eosinophilic globules of immunoglobulin within plasma cells) may be prominent, but lymphoepithelial lesions, amyloid and Dutcher bodies should be absent. Central scarring is frequent (fig 6C). Lymphangitic spread of lymphocytes into the perivascular interstitium surrounding the lesion may be present, but it is focal and mild. Uncommonly, giant cells may be seen. Plaque-like infiltration of the pleura and bronchial cartilage invasion are typically absent.1 Reactive lymphadenopathy may be seen in adjacent hilar, mediastinal, or paraoesophageal lymph nodes.

Nodular lymphoid hyperplasia. (A) A nodular aggregate of lymphoid tissue containing germinal centres and areas of fibrosis. (B) This area shows a dense lymphoid infiltrate with germinal centres and a dense fibrous scar. (C) The dense fibrous scar is infiltrated with lymphocytes and plasma cells.
Immunohistochemical analysis shows a mixture of B and T cells with germinal centres expressing CD20 (B cells) and the interfollicular lymphocytes expressing CD3 (T cells), CD43 (leucocytes except resting B cells), and CD5 (T cells, subset of B cells).1 A polyclonal pattern of reactivity is seen with immunohistochemical staining for κ and λ immunoglobulin light chains. Bcl-2 staining is absent in the germinal centres and coexpression of CD20 with CD43 or CD5 is not seen. No rearrangements of the immunoglobulin heavy chain gene should be found by molecular analysis using the polymerase chain reaction.1
DIFFERENTIAL DIAGNOSIS
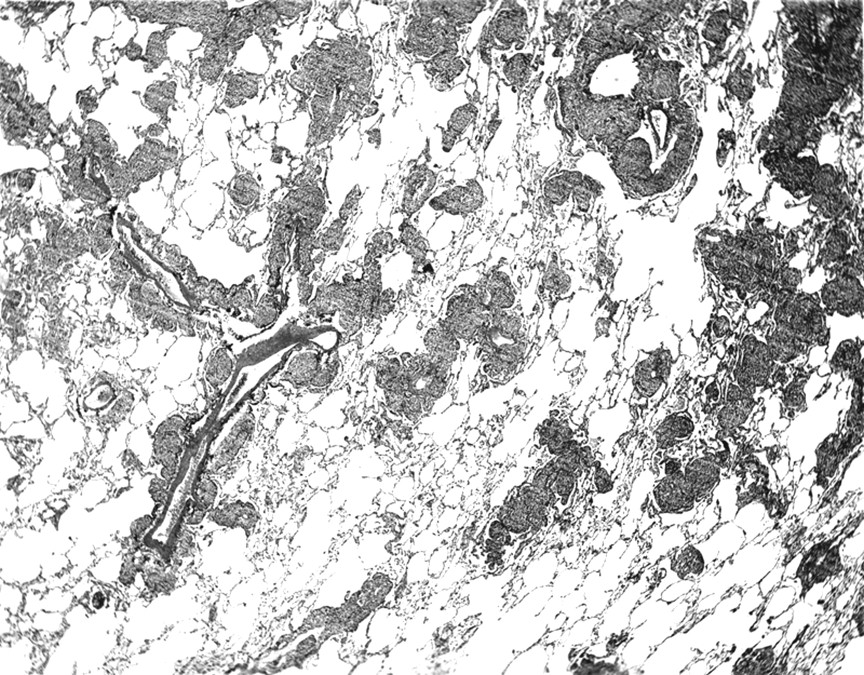
MALT lymphoma is the most difficult lesion to be separated from nodular lymphoid hyperplasia.1 Other differential diagnoses include inflammatory pseudotumour,49plasmacytoma,50 lymphocytic interstitial pneumonia, and follicular bronchiolitis. The major criteria for distinction of MALT lymphoma from nodular lymphoid hyperplasia are summarised in table 1. MALT lymphoma is relatively common compared with nodular lymphoid hyperplasia, which is rare.51 For this reason it is wise to be cautious before suggesting a nodular pulmonary lymphoid lesion is benign. MALT lymphomas and nodular lymphoid hyperplasia have many overlapping clinical and radiological features. The presence of multiple bilateral pulmonary nodules should increase the suspicion for a lymphoma. Histologically, MALT lymphomas often have reactive germinal centres and large mantle zones may be present. Follicular colonisation (infiltration of lymphoid follicles by the malignant B cells of the MALT lymphoma) may be seen. Other features that favour a MALT lymphoma include conspicuous infiltrative growth with prominent lymphangitic spread (fig 7), plaque-like infiltration of the pleura, and invasion of bronchial cartilage.1 ,51 Lymphoepithelial lesions are seen in MALT lymphomas but are not a prominent feature in nodular lymphoid hyperplasia. Monocytoid B cells are often seen in MALT lymphomas and plasma cells may be prominent, while these features are usually not conspicuous in nodular lymphoid hyperplasia. Immunohistochemical analysis for immunoglobulin light chains shows a monoclonal pattern in approximately 40% of MALT lymphomas,1 ,51 while a polyclonal pattern should be seen in nodular lymphoid hyperplasia. Rearrangement of the immunoglobulin heavy chain gene is seen in the majority of MALT lymphomas, but should be absent in nodular lymphoid hyperplasia.1 ,51
Nodular lymphoid hyperplasia versus malt lymphoma
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MALT lymphoma. This dense lymphoid infiltrate shows the pattern of “tracking” along lymphatic routes, a feature characteristic of pulmonary MALT or small lymphocytic lymphoma. The lymphoid cells are infiltrating around the bronchovascular bundles in the region of the lymphatic spaces.
Nodular lymphoid hyperplasia differs from lymphocytic interstitial pneumonia and follicular bronchiolitis in that the latter two both affect the lung diffusely and do not form a discrete nodular mass.1 Inflammatory pseudotumour embraces a spectrum of lesions ranging from a plasma cell granuloma to a fibrohistiocytic morphology.48 The morphology of the plasma cell granuloma consists of a mixture of lymphocytes and plasma cells with mild to moderate numbers of spindle cells with a fibroblastic or myofibroblastic appearance. In the fibrohistiocytic variant the lesion consists mostly of spindle-shaped cells with relatively sparse inflammatory infiltrates. While dense scarring may be present in nodular lymphoid hyperplasia, the presence of a proliferation of fibrohistiocytic or myofibroblastic spindle cells is not seen. Plasmacytoma also differs in that it consists of a pure population of plasma cells that show a monoclonal pattern with immunohistochemistry for immunoglobulin light chains.50