Article Text

Statistics from Altmetric.com
For hundreds of years salicylates have been used to treat fever and pain. Following the discovery that aspirin inhibits the biosynthesis of prostanoids,1 particularly thromboxane A2 (TXA2) in blood platelets, aspirin gained a new clinical indication as an antiplatelet and antithrombotic drug. Aspirin acetylates the serine 529 residue of cyclo-oxygenase 1 (COX-1) in platelets and megakaryocytes2; however, tyrosyl residues in inducible COX-2 are also acetylated.3 The anti-inflammatory action of aspirin depends on its interaction with COX-2 and subsequent removal of proinflammatory prostanoids or, possibly, on the appearance of cytoprotective 15-epi-lipoxins.4 Only diclofenac induced COX-2 remains insensitive to aspirin.5 Inhibition of nuclear factor kappa B (NFκB) activation is another mechanism of anti-inflammatory action of salicylates.6 Many unusual actions of aspirin are associated with its acetylating power that extends beyond serine residues in COX-1 or tyrosine residues in COX-2. For instance, the anti-cataract effect of aspirin seems to be associated with acetylation of cysteinyl residues of lens γ-crystallins which prevents the formation of opaque disulphide bonding.7 Aspirin affects the rheological properties of erythrocytes,8 decreases erythrocyte mediated activation of platelets,9 and modifies the functioning of haemoglobin by acetylation of its lysyl residues.10 Even platelet membranes possess protein sites available for acetylation by aspirin.11 In humans thrombinogenesis is inhibited by aspirin, possibly as a consequence of acetylation of either platelet membranes or active sites of prothrombin.12 The relation between sodium salicylate and aspirin is complex. Protective effects of sodium salicylate against inhibition of COX by aspirin have been reported in many systems including patients with aspirin induced asthma.13 On the other hand, both drugs enhance the generation of nitric oxide by activated murine macrophages14 or by cultured rat smooth muscle cells.15
The antithrombotic activity of aspirin is indicated, firstly, by inhibition of TXA2 generation in platelets and, secondly, by inhibition of plasma thrombinogenesis through an unknown mechanism.12 The effects of aspirin on fibrinolysis and thrombolysis are even more complex.16 Here, we describe in vivo mechanisms of thrombolytic interactions between aspirin and another powerful antiplatelet agent, ticlopidine,2 which has endothelium mediated thrombolytic properties of its own.17 ,18 We also studied the activity of two other thienopyridines, clopidogrel and its enantiomer.
Methods
THROMBOLYSIS ASSAY EX VIVO
Wistar rats weighing 300–350 g were anaesthetised with thiopentone (30 mg/kg ip) and injected intravenously with unfractionated heparin (800 units/kg) and half an hour later extracorporeal circulation between the left carotid artery and jugular vein was established. On its way arterial blood superfused a collagen strip from rabbit Achilles tendon (1.5 ml/min). Its gain or loss in weight was continuously monitored by an auxotonic Harvard transducer.17 The strip gained in weight by 80–120 mg during the first 20 minutes of superfusion and its weight stayed unchanged during the next 3–5 hours of the experiment owing to the deposition of thrombi consisting of activated platelets, blood cells, and fibrin.19 Arterial blood pressure was continuously recorded from the right carotid artery. In our system dispersion of thrombi, as indicated by a loss in weight of the superfused strip, occurred after intravenous injections of prostacyclin or iloprost (0.03–1.0 μg/kg), glyceryl trinitrate (30–100 μg/kg), methacholine hydrochloride (3–10 μg/kg), or bradykinin (0.1–1.0 μg/kg). Streptokinase (3–30 megaunits/kg) produced a biphasic thrombogenic/thrombolytic response.17 ,19 We have studied the thrombolytic effects of aspirin (ASA, 0.1–100 mg/kg) and the antiplatelet thienopyridines ticlopidine and clopidogrel (SR25990C) in doses of 1–30 mg/kg, as well as the R enantiomer of clopidogrel deprived of antiplatelet properties (SR25989C).
6-KETO PGF1αAND t-PA ASSAY
In parallel to thrombolysis, we also assayed 6-keto-prostaglandin F1α (PGF1α) (enzyme immunoassay (EIA) kit, Cayman Chemical Co, Ann Arbor, MI, USA) and tissue plasminogen activator (t-PA) (EIA kit, Biopool TintElize t-PA antigen, Umea, Sweden). For the 6-keto-PGF1α EIA whole blood samples (250 μl) were collected in Eppendorff tubes with indomethacin (final concentration 10 μM). For the t-PA assay plasma samples (250 μl) were collected. Samples were stored at –70°C for a maximum of one week, centrifuged at 400g, and assayed according to the manufacturer's protocols. Thienopyridines were kindly donated by Sanofi Recherche (Toulouse, France). Lysyl aspirin and other reagents were purchased from the Sigma Chemical Co (St Louis, MO, USA).
STATISTICAL ANALYSIS
Arithmetical means with standard error (SE) are presented. The data were analysed by two way analysis of variance (ANOVA). Statistics were analysed using GraphPad Software for Windows.
Results
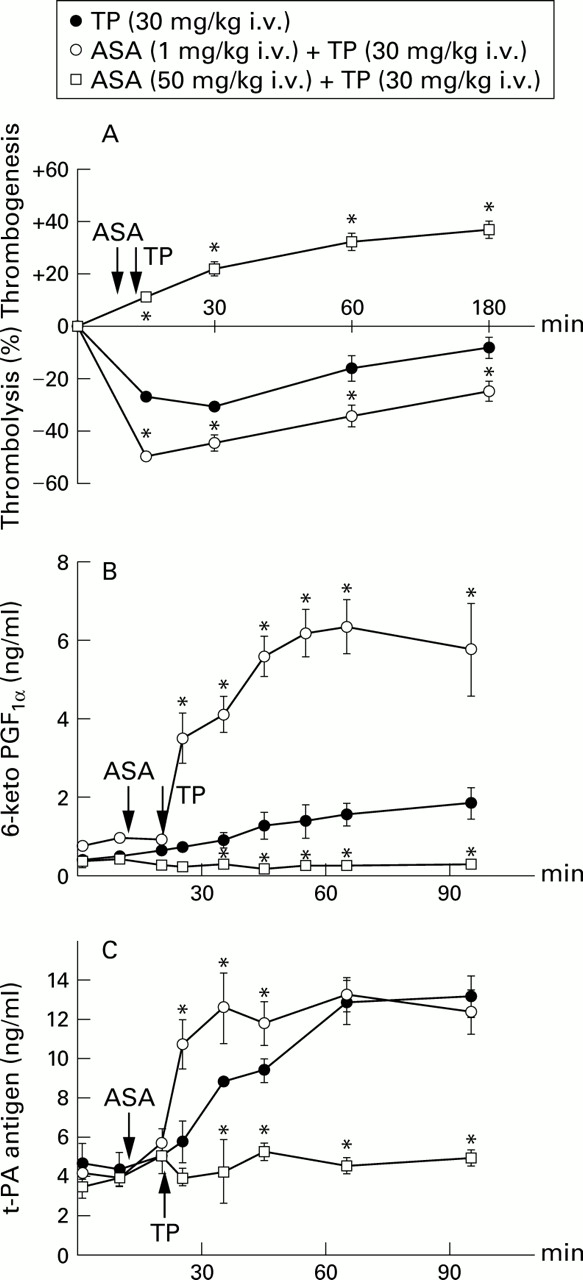
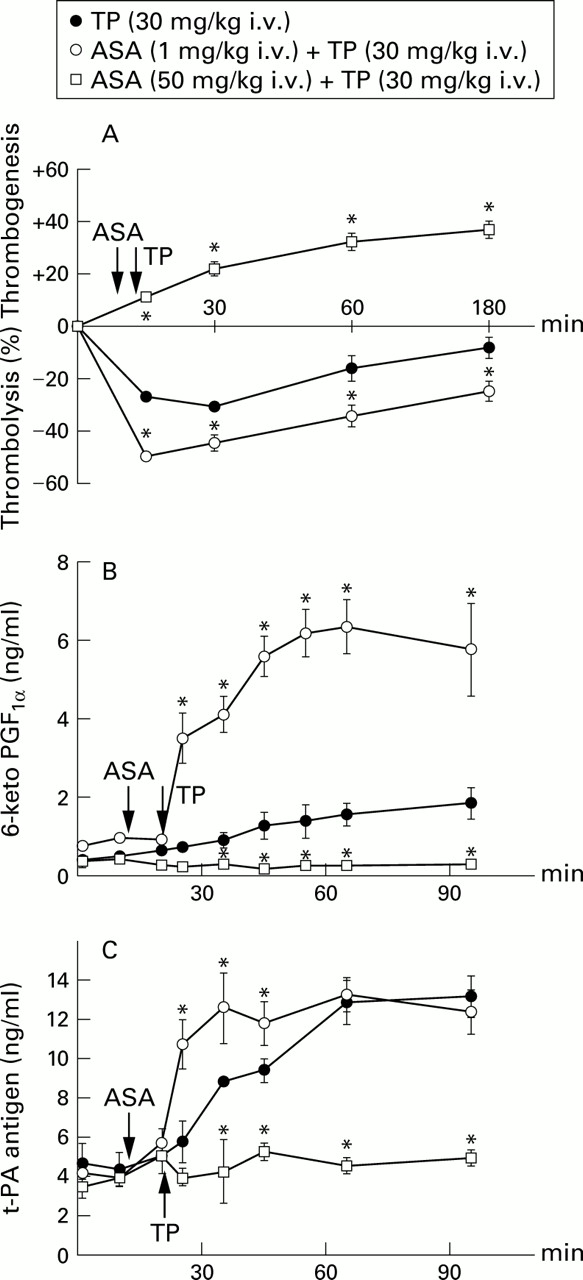
At doses of 0.1 mg/kg (n = 3) and 5–50 mg/kg (n = 12) ASA had no thrombolytic effect. At doses of 0.3, 1.0 and 3.0 mg/kg ASA produced a transient (15–30 minutes duration) thrombolytic response, with maximum thrombolysis being evoked at 1 mg/kg (11.8 (3.2)%, n = 5). Unlike ASA, the thienopyridines ticlopidine, clopidogrel, and the R enantiomer of clopidogrel produced dose dependent thrombolysis in a range of doses from 3 to 30 mg/kg. Ticlopidine (30 mg/kg), clopidogrel (10 mg/kg), and the R enantiomer of clopidogrel (10 mg/kg) had similar thrombolytic potency, reducing the weight of thrombi by 30.5 (1.8)% (n = 15), 28.3 (2.3)% (n = 9), and 30.4 (1.9)% (n = 8), respectively (fig 1). The thrombolytic response to ticlopidine (fig 2A) was associated with a progressive increase in blood levels of 6-keto-PGF1α (fig 2B) and t-PA (fig 2C). Pretreatment with a low dose of ASA (1 mg/kg) significantly augmented not only the thrombolytic response to ticlopidine (fig 2A) but it also dramatically increased the release of 6-keto-PGF1α and t-PA (fig 2B and C). In contrast, ASA at a high dose of 50 mg/kg abolished ticlopidine induced release of both endothelial products and, at the same time, it reversed the action of ticlopidine from thrombolytic to thrombogenic (fig2).
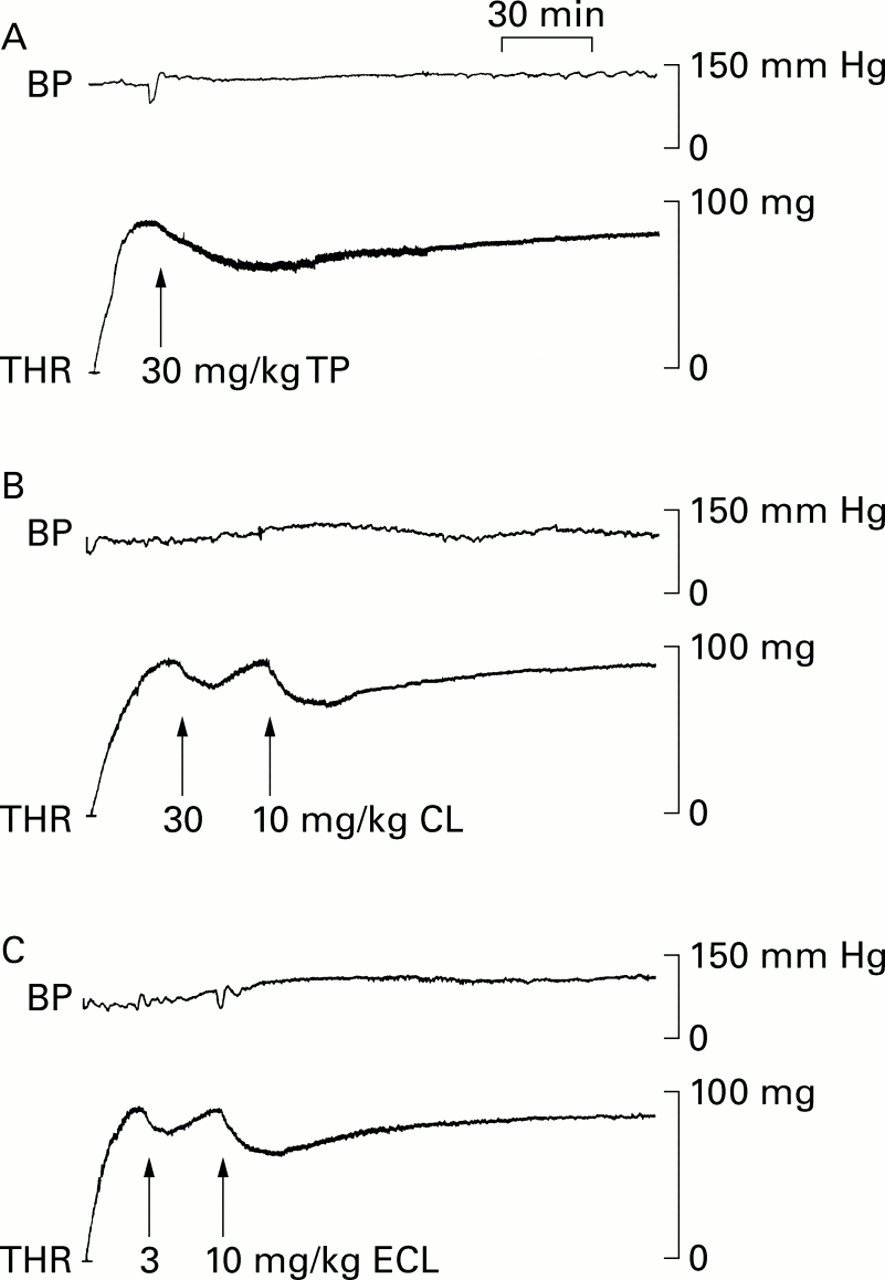
Original tracings of thrombolytic (THR) and blood pressure (BP) effects of the thienopyridines ticlopidine (TP), clopidogrel (CP), and the R enantiomer of clopidogrel (ECL). The tracings derive from three anaesthetised Wistar rats (A, B, and C) with thrombi superfused with arterial blood in extracorporeal circulation.
{kind=link}
{kind=link}
Effects of pretreatment with low dose (○) and high dose (□) aspirin (ASA) on (A) thrombotic potency, (B) blood level of 6-keto-PGF1α, and (C) plasma level of t-PA antigen by ticlopidine (TP, ●). Each point represents the arithmetical mean of six experiments in anaesthetised Wistar rats with extracorporeal circulation with blood superfused thrombi. Vertical bars represent two standard errors of mean. *P<0.05 (○ vs ● or □ vs ●).
Discussion
In our model drugs may produce thrombolysis by disaggregation of platelet clots, by fibrinolysis, or by both mechanisms. The capacity of aspirin to acetylate proteins, including serine residues in platelet COX-1,2 ,3 ,11 ,12 ,19 seems to be the dominant mechanism for its antithrombotic action. Aspirin acted as a weak and short acting thrombolytic agent in our model within a narrow range of doses (0.3–3.0 mg/kg). Unlike aspirin, unstable metabolites of thienopyridines (ticlopidine and clopidogrel) are supposed to antagonise platelet ADP receptors, and the antiplatelet action of ticlopidine20 and clopidogrel21 is potentiated by aspirin. However, ticlopidine also shows a distinct thrombolytic action in humans, cats,17 and rats which is independent of platelets.18 In vitro, ticlopidine, clopidogrel and the R enantiomer of clopidogrel release PGI2 and nitric oxide (NO) from cultured human and bovine endothelial cells.22 In our study we found that the thrombolytic response to ticlopidine in rats was accompanied by an increase in blood levels of 6-keto-PGF1α (a product of decomposition of PGI2) and of t-PA antigen. Release of these endothelial products might be responsible for the disaggregatory and fibrinolytic actions of ticlopidine. This assumption was supported by the fact that pretreatment with aspirin at a high dose of 50 mg/kg not only prevented ticlopidine from releasing 6-keto-PGF1α and t-PA (presumably from vascular endothelium), but also converted the thrombolytic action of ticlopidine to a thrombogenic one. On the other hand, pretreatment with aspirin at a low dose of 1 mg/kg increased the thrombolytic potency of ticlopidine and enhanced its ability to release 6-keto-PGF1α and t-PA antigen. The low dose of aspirin clearly did not prevent the secretory activity of the endothelium but, on the contrary, promoted it, possibly by selective removal of TXA2 from interacting with endothelium.
In conclusion, aspirin not only potentiates the disaggregatory and antithrombotic action of ticlopidine at the platelet level, as reported by others,20 but at low doses it also enhances the endothelium mediated thrombolytic potency of ticlopidine as indicated by a rise in blood levels of PGI2 and t-PA. The extraplatelet mechanism of the thrombolytic action of thienopyridines is suggested by the fact that clopidogrel (an antiplatelet agent) and its enantiomer (deprived of antiplatelet activity) were both equally as potent as thrombolytic agents in our in vivo model. We therefore assume that thienopyridine molecules have two pharmacological properties that are independent of each other: (a) antiplatelet potential that is revealed during biotransformation to an active and unstable ADP receptor antagonist, and (b) thrombolytic potential associated with a native chemical moiety that stimulates the secretory function of the vascular endothelium. Aspirin modifies both these pharmacological properties of thienopyridines in a dose dependent manner.
Acknowledgments
This research was funded by State Committee for Scientific Research.