Article Text

Statistics from Altmetric.com
Hereditary haemorrhagic telangiectasia (HHT, Rendu-Osler-Weber syndrome) exemplifies an important group of diseases which have catalysed advances in the understanding of fundamental pathophysiological mechanisms. In this paper areas of clinical management of HHT are discussed and the molecular pathogenesis is reviewed. The first section is aimed at all clinicians and concentrates on the recognition of a disorder in which silent cerebral and pulmonary involvement may be life threatening if left untreated. Recent data concerning the diagnostic and treatment modalities for pulmonary arteriovenous malformations (PAVMs) are also reviewed, and the growing concern that many patients with HHT may have small or residual PAVMs is highlighted. The paucity of good longitudinal data on these patients and others with different forms of HHT highlights the need for further clinical studies. In the second section the results of molecular research which suggests a role for receptors and ligands of the transforming growth factor (TGF)-β superfamily in the pathogenesis of this vascular disease are discussed. The means by which such information may relate to the clinical heterogeneity observed in HHT are specifically addressed, and more fundamental questions such as how reduced cell surface expression of endoglin predisposes a patient to develop PAVMs are also discussed.
Hereditary haemorrhagic telangiectasia
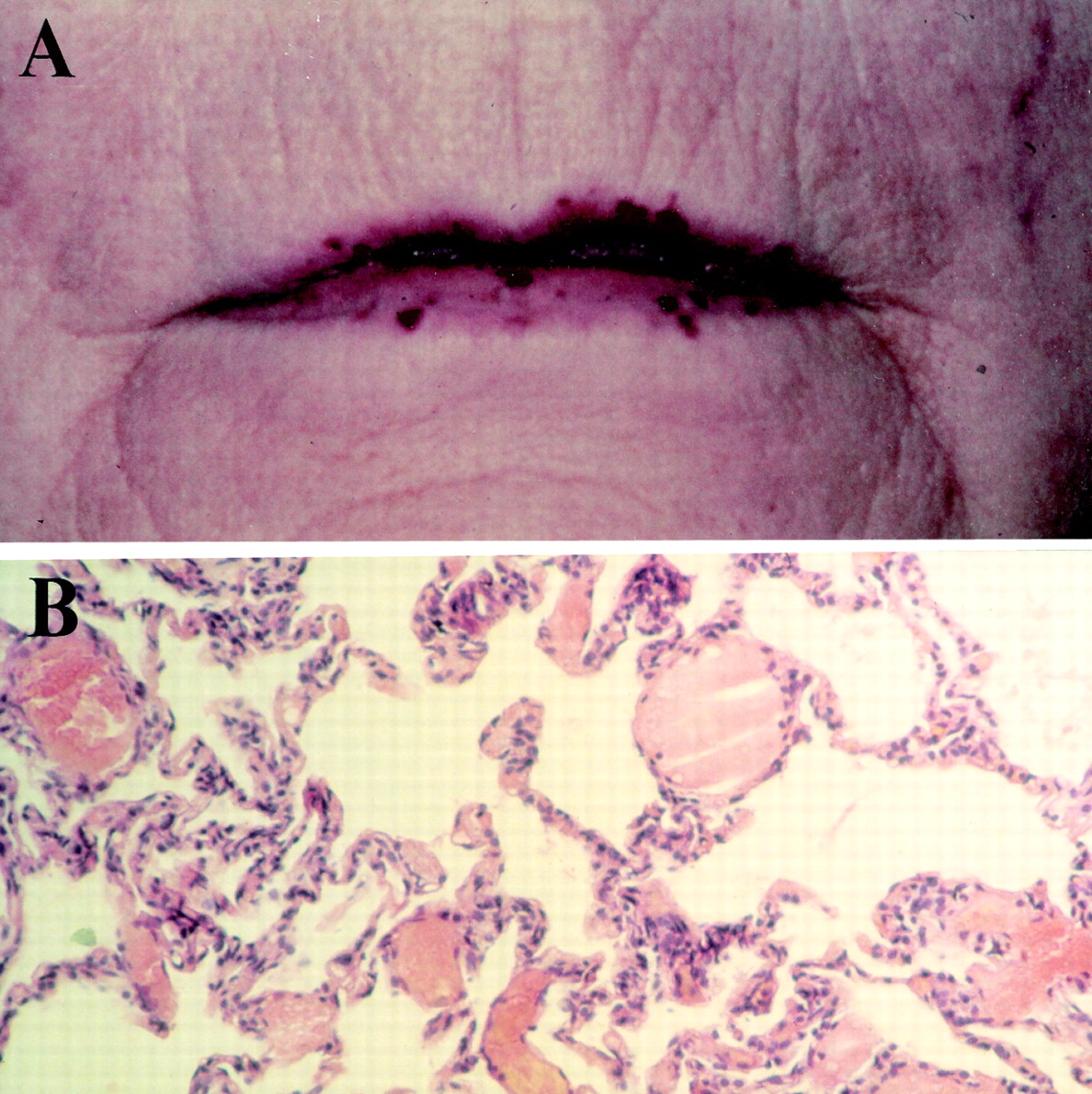
The classical patient with the vascular disorder hereditary haemorrhagic telangiectasia (HHT) has nose bleeds, dilated blood vessels over the lips and finger tips, and gastrointestinal bleeding in later life. However, this clinical scenario represents only one of the presentation patterns of HHT.1 ,2 It is now recognised that, in addition to microscopic mucocutaneous telangiectases derived from post capillary venules (fig 1A),3 HHT leads to the development of larger abnormal vascular structures at other sites. Arteriovenous malformations in the pulmonary, cerebral, and hepatic circulations account for some of the most devastating clinical complications of the disease.

Hereditary haemorrhagic telangiectasia (HHT). (A) Mucocutaneous telangiectasia on the lips of a patient with HHT. In these simple telangiectases, thin walled endothelial cell lined vessels resembling dilated post capillary venules connect apparently normal capillaries and draining venules, with a high frequency of direct arteriovenous communications.3 ,6 (B) Histology of microscopic PAVMs. Haemotoxylin and eosin stained slide taken from an open lung biopsy specimen displaying dilated vascular structures in place of normal corner vessels. These were not visible radiologically, although they caused hypoxaemia and a right-to-left shunt.7 As in mucocutaneous telangiectases,3it is thought that larger lesions (see fig 2) may arise from microscopic PAVMs by a process of remodelling.
The autosomal dominant inheritance pattern of HHT has enabled identification of the underlying genetic defects, prompting increased scientific interest in the disorder. Mutations in at least two genes have been shown to be associated with HHT in different families:endoglin on chromosome 9,4 andALK-1 (activin receptor-like kinase 1) on chromosome 12.5 Both genes encode endothelial cell transmembrane proteins that can be defined as components of the receptor complexes for growth factors of the TGF-β superfamily. This suggests that disease pathogenesis is likely to result from perturbation of physiological effect(s) of these growth factors in vascular development or homeostasis. At the present time, the factors implicated and the mechanisms which regulate their action remain speculative. Relevant data are discussed further in the final section of this review, to which the non-clinical reader is referred directly.
CLINICAL ASPECTS OF HHT
HHT is more common than previously appreciated, with prevalence rates exceeding one in 10 000 in some regions.8-11 The disease displays age related penetrance, with manifestations developing throughout life and varying between affected individuals, even individuals from the same family. Heterozygotes account almost exclusively for the patient population: there are very few reports of probable homozygous cases.12-14
The common clinical manifestations of HHT are summarised in table 1which also provides an overview of the presentation patterns and treatments for the manifestations that are usually managed by appropriate specialists (epistaxis, mucocutaneous telangiectasia, and gastrointestinal lesions); further information may be found in excellent recent reviews.1 ,2 A significant proportion of patients with HHT have pulmonary and cerebral vascular involvement. These manifestations differ from other common sites of involvement since silent lesions may cause considerable morbidity and mortality if left untreated.
Clinical features of HHT
The key to appropriate management of patients with HHT is to be alert to the possibility of additional visceral involvement and hence the importance of establishing a diagnosis. This point needs to be considered by the physician as individuals presenting with HHT are often unaware that they have a familial disease. Current clinical diagnostic criteria require the presence of three out of four key features for a definitive diagnosis—namely, spontaneous recurrent epistaxis, telangiectases at characteristic sites, a visceral manifestation, and an affected first degree relative.40 To reduce the number of cases overlooked and deprived of suitable screening regimes, the label of “suspected HHT” should be used if two features are present, and particularly in the presence of PAVMs which are rare in patients without HHT.40 Since HHT may present to a number of clinical specialities, the significance of a particular presentation is often overlooked.41
CEREBROVASCULAR MALFORMATIONS AND HHT
Cerebral manifestations including telangiectases, venous malformations, and arteriovenous malformations (CAVMs) are under-recognised in patients with HHT. Cerebral involvement is usually said to affect 5–10% of patients with HHT,1 ,8 but a much higher incidence is seen when asymptomatic patients are screened.42
The highest complication rate is observed in high flow CAVMs which may present with headache, epilepsy, ischaemia (due to a vascular steal effect), or haemorrhage. Symptomatic lesions may be treated by microsurgical excision, stereotactic radiotherapy for lesions less than 3 cm in diameter, and embolisation.1 ,2 There are no trials comparing embolisation with other forms of treatment but a recent review suggests that patients offered stereotactic radiotherapy fared less well in terms of immediate mortality, obliteration of the lesion, and post-intervention neurological deficits than patients treated by microsurgery.43
There has been considerable debate about the optimal therapy for asymptomatic CAVMs. The natural history of HHT associated CAVMs is not entirely clear though it is usually assumed to be equivalent to non-HHT CAVMs. A risk of haemorrhage of 2% per annum, varying with certain features of the lesion,44 ,45 is generally used as the basis of careful risk-benefit analyses. These analyses suggest that in asymptomatic patients the risks of haemorrhage with expectant treatment outweigh the risks of intervention, particularly in young patients.46 However, the risk assessment for individual patients must consider the patient’s age, specific features of the lesion, and particularly the available therapeutic expertise.2 ,46
Since intervention may be recommended for asymptomatic patients, some centres offer screening programmes for families with HHT using intravenous digital subtraction angiography (DSA).2 ,47This avoids the morbidity from conventional cerebral angiography although the limitations of this relatively non-invasive technique have to be recognised.
Pulmonary arteriovenous malformations and HHT
OVERVIEW
More than 20% of patients with HHT develop pulmonary arteriovenous malformations (PAVMs) which range from diffuse telangiectases (fig 1B)48-50 to large complex structures consisting of a bulbous aneurysmal sac between dilated feeding arteries and draining veins (fig 2).51 Around 95% of feeding arteries come from the pulmonary rather than systemic circulation.50 Since approximately 70% of PAVMs occur in patients with HHT,52-54 their detection should prompt a thorough review of the patient and his or her family. Multiple lesions are particularly suggestive of an association with HHT.52 ,53 ,55
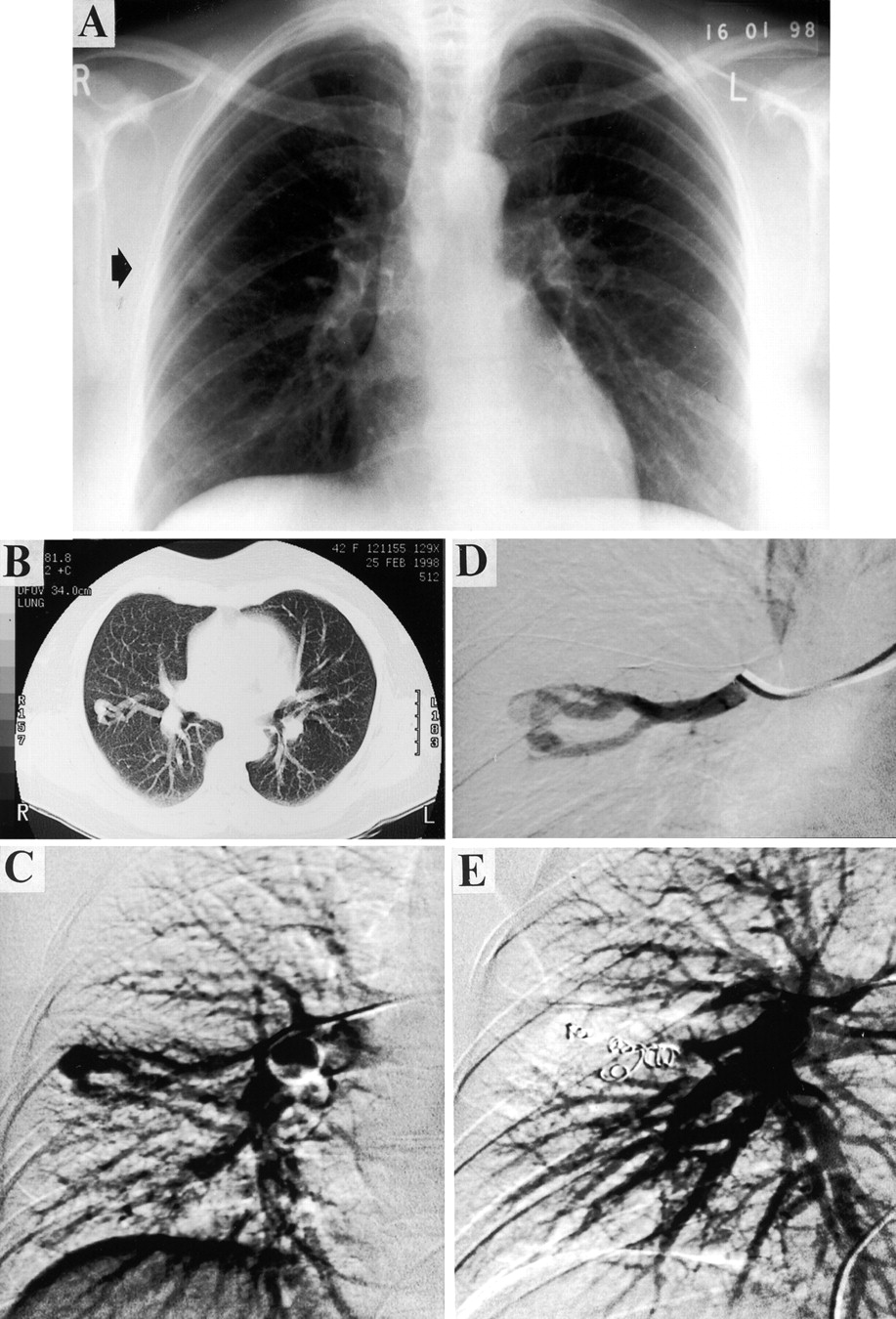
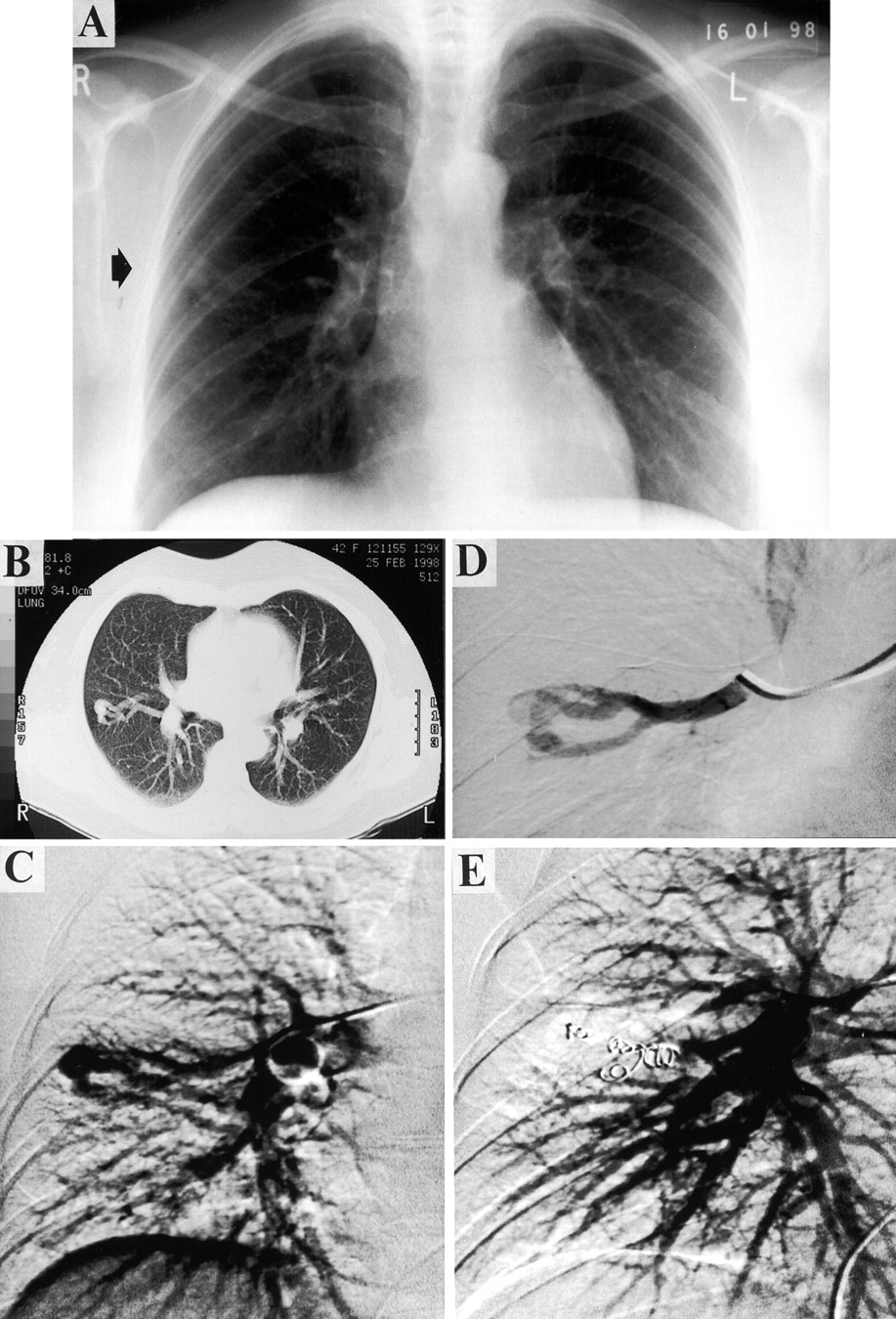
A PAVM from diagnosis to treatment. A 42 year old woman presented with lethargy and a history of previous nose bleeds. A chest radiograph (A) and CT scan (B) suggested the presence of a PAVM. She was marginally hypoxaemic (Pao 2 11.4 kPa on air) with no orthodeoxia. Her right-to-left shunt measured by 100% oxygen was 6–8%. Angiography confirmed a single lesion in the right mid zone (C). Selective angiography (D) revealed a complex structure with three feeding arteries and a single draining vein. The feeding vessels were embolised with five metallic coils (E). Two months after embolisation there was significant improvement in oxygenation (Pao 2 13.5 kPa).
PAVMs tend to increase in size,50 ,56 ,57 especially if multiple,56 and rarely regress spontaneously.57 The mortality rate in historical reviews of untreated but usually symptomatic patients with PAVMs over periods of 15 years or less ranges from 4% to 22%52 ,55 ,56 ,58 ,59 and, in severe cases, up to 40%.60 Complications are more common when HHT is present.61 The abnormal vessels may bleed into a bronchus or the pleural cavity, sometimes with a fatal outcome.60 ,62 ,63 However, it is the functional consequences of the direct communications between pulmonary and systemic circulations, bypassing the capillary bed, that most commonly cause problems. Such right-to-left shunts cause hypoxaemia and the absence of a filtering capillary bed allows paradoxical embolism of particulate matter which can reach the systemic arteries, causing clinical sequelae, particularly in the cerebral circulation.
These processes account for the clinical features on presentation (table 2). It should be noted that dyspnoea was common (47%), though many patients tolerate hypoxaemia—even exercise aggravated hypoxaemia—well, reflecting their low pulmonary vascular resistance and ability to generate supranormal cardiac outputs64-66which may increase further on exercise.67 Haemoptysis was seen in 11%. Up to 50% of patients had no respiratory complaints on presentation despite physical signs (such as cyanosis, clubbing, or a vascular bruit) or an abnormal chest radiograph. Most importantly, many patients had minimal respiratory symptoms when presenting with neurological complications of PAVMs (transient ischaemic attacks, strokes and cerebral abscesses).52 ,55 PAVMs accounted for two thirds of HHT-related neurological presentations in one series68 and they carry a significant mortality.69
Symptoms and signs of PAVMs at presentation in major series. Many patients will have symptoms, stigmata or a family history of HHT
It is recommended that patients with PAVMs should receive antibiotic prophylaxis prior to dental and surgical interventions to reduce embolic abscesses,83-85 although there is no direct evidence of benefit. Formal treatment of PAVMs by periangiographic embolisation techniques or surgery is required to alter radically the long term prognosis for patients with PAVMs.
DIAGNOSIS OF PAVM
Pulmonary angiography is required for therapeutic embolisation and is also mandatory to determine the position and structure of abnormal vascular lesions prior to surgical treatment. Angiography is labour, cost and radiation intensive and its use should be limited to individuals in whom non-invasive diagnostic tests strongly suggest the presence of PAVMs. The methods used depend upon local experience but several recently published studies are worth reviewing.
Initial investigations
Chest radiographs
Moderate sized PAVMs appear as rounded, well circumscribed lesions (fig 2), often with associated band shaped shadows resulting from dilated feeding and draining vessels. The intensity of shadowing may be diminished or enhanced respectively by the Valsalva and Muller manoeuvres.86 Patients with PAVMs often present with an abnormality on the chest radiograph,55 ,57 and this may have led to an overestimate of the frequency of radiographic abnormalities at presentation; it is now recognised that a normal posterior-anterior and lateral chest radiograph does not rule out PAVMs (table 3),42 ,56 ,87 particularly in patients with small or diffuse malformations.
Comparison of findings from different methods used to detect PAVMs
Assessment of hypoxaemia—Unexplained and often profound hypoxaemia is the hallmark of large PAVMs, but there are additional features that may help to establish more specific diagnostic tests. Further desaturation on assuming the upright posture, orthodeoxia, is common in patients with PAVMs,91 due primarily to a gravity induced increase in flow through basally situated shunts (approximately 70% of PAVMs),50 ,52 ,53 ,58 ,71 ,75 which increases the right-to-left shunt.92 Data on the effect of exercise on shunt flow and hypoxaemia are contradictory.73 ,93
The detection of hypoxaemia lacks specificity as a diagnostic test (table 3)42 but it can identify patients worthy of further investigation. In 66 patients who had undergone embolisation, the presence and extent of residual PAVM disease was related to oxygen saturation (despite the imprecision of pulse oximetry94 ,95) with the patients erect and supine and on maximal exercise, and to the change in oxygen saturation between being erect and supine.89 Oxygen saturation with the patient erect was the best predictor of the presence or absence of disease, though sensitivity and specificity were again too low to recommend it as the sole diagnostic screening test (table 3).
Confirmatory studies
Abnormal architecture
Helical CT scanning with three-dimensional reconstructions conveniently identifies small, multiple lesions; it can also identify thrombosed88 ,96 and, with contrast, recanalised structures.77 It exposes patients at risk of recurrent disease to a significant dose of radiation, and misdiagnoses have been reported.97 At present NMR screening is less effective than computed tomographic (CT) scanning or pulmonary angiography as small PAVMs with rapid blood flow are not visualised,98but methodology is improving.
Intravenous digital subtraction angiography (DSA) of pulmonary arteries is performed prior to formal catheter pulmonary angiography in some centres to visualise the pulmonary vasculature, particularly as part of an outpatient screening programme.42 However, this method is less likely to detect certain lesions than formal angiographic studies.42
Detection of right-to-left shunts—In normal individuals the right-to-left shunt is less than 2% of the cardiac output99 ,100 and ascribed to “post-pulmonary” shunting due to the mixing of pulmonary venous blood with deoxygenated blood from bronchial, mediastinal, and thebesian veins. The flow through right-to-left shunts is usually expressed as a fraction of total flow (Q˙s/Q˙T) and may be calculated from the reduction in arterial oxygenation or by anatomical methods using particles 7–11 mm in diameter which normally impact in pulmonary capillaries but pass through large calibre shunt vessels.
100% inspired oxygen breathing method: Calculating the shunt from the arterial oxygen tension after breathing 100% oxygen for 20 minutes has been considered the gold standard for non-invasive methods of estimating the size of the shunt.99 Effects of ventilation-perfusion inequalities should be overcome since blood derived from poorly ventilated alveoli will be fully saturated, though it should be noted that this method will also detect post-pulmonary shunting. The anatomical intrapulmonary shunt may be underestimated if microscopic arteriovenous malformations are participating in gas exchange,101 although such shunts may not be treatable. Disadvantages include the requirements for a mask and bag to give 100% oxygen, arterial blood gas sampling and difficulties in calibrating the oxygen electrode for high oxygen tensions using blood tonometry or commercially available sealed buffer solutions. In the absence of right heart catheterisation mixed venous oxygen content is usually estimated. The normal arteriovenous oxygen content (A–Vo 2) difference is 5 ml/100 ml. Recognising that patients with an A–V shunt are likely to have an increased cardiac output, and that a lower A–Vo 2 difference of 3.5/100 ml may be more appropriate for this situation, some centres calculate the shunt for both normal and high cardiac outputs (using A–Vo 2 differences of 5 ml/100 ml and 3.5/100 ml, respectively), with the final shunt fraction given as a range between the two values (as in fig 2). Using 100% inspired oxygen, a right-to-left shunt of more than 5% is considered abnormal.
Radionuclide scanning: following intravenous injection of technetium-99m (99mTc)-labelled albumin microspheres or macroaggregates, the right-to-left shunt can be calculated by comparing the quantity reaching the systemic circulation with the total quantity received.102 ,103 Shunts of up to 3.5% are detected in normal subjects.102 Recent data from one institution with extensive experience using this method indicated that, in patients who had undergone embolisation of PAVMs, a shunt measurement of >3.5% had 87% sensitivity and 61% specificity for the presence of residual disease.89 However, suitable facilities are not always available.
Contrast echocardiography can be used to assess the presence of right-to-left shunting, although currently the shunt cannot be quantified by this method. Microbubbles generated by intravenously injected echocontrast should be lost on passage through the normal pulmonary capillary bed; the appearance of echoes in the left ventricle indicates the presence of a right-to-left shunt, a delay of 2–5 seconds indicating an intrapulmonary shunt rather than an intracardiac shunt in which left ventricular echoes appear almost instantly.104 Contrast echocardiography may be too sensitive for clinical use, however (see below).
Comparisons of non-invasive screening procedures and correlation with angiographic confirmation of PAVMs
Non-invasive methods have rarely been compared, although an excellent correlation was obtained for right-to-left shunt measurements obtained by the 100% inspired oxygen and radioisotope methods in one centre,103 and between 100% oxygen and the multiple inert gas elimination technique (MIGET) in another, though only when right heart catheter data allowed appropriately high mixed venous oxygen content values to be used in the shunt equation for 100% oxygen.65 Contrast echocardiography was more sensitive than measurement of arterial blood gas tensions and chest radiography.105 Indirect comparisons between PAVM screens in neighbouring European HHT populations indicate that contrast echocardiography is also more sensitive than 100% inspired oxygen methods; contrast echocardiography generated a high diagnostic yield of 51% (many of whom did not have angiographically detectable AVMs90) compared with 33% in a separate HHT population screened by 100% oxygen methods.42 It has therefore been suggested that contrast echocardiography may be an excessively sensitive technique, and this needs to be determined.
All non-invasive methods occasionally fail to detect PAVMs which are subsequently diagnosed by angiography (see table 3). More commonly the inverse is seen; an abnormally high shunt is detected by non-invasive methods but no shunt is seen at formal pulmonary artery catheter angiography. Nearly 40% of a total of 109 PAVMs identified by CT scanning were not detected at angiography88 and an abnormal transfer factor for carbon monoxide (Tlco), observed in a proportion of patients with PAVMs, often persists following apparently successful embolisation therapy.73 ,75 It is presumed that lesions missed by formal pulmonary angiography (or responsible for the reduced transfer factors) were too small for detection (as in fig 1B) and will not be amenable to embolisation. When widespread they cause profound hypoxaemia,7 ,48 ,49 ,54 but the clinical significance of smaller numbers of such PAVMs is unknown.
Conclusions and recommendations
Patients considered at risk of PAVMs because of suspicious symptoms, signs or radiological appearances should be investigated with at least measurement of arterial blood gas tensions and/or supine and erect oximetry, together with posterior-anterior and lateral chest radiographs. If there is still concern, at least one non-invasive method to assess the presence of PAVMs or degree of right-to-left shunting should be undertaken before formal pulmonary angiography is warranted; the choice of procedure is still likely to depend upon local experience. Shunt measurements are likely to be performed by 100% oxygen methods in respiratory units, nucleotide scanning in centres where facilities are available, and elsewhere by contrast echocardiography noting that this technique is likely to overestimate the number of patients with treatable lesions. When a shunt is detected but pulmonary angiography proves negative, the possibility of intracardiac shunting may need to be excluded by echocardiography.
Screening programmes to detect PAVMs in patients with HHT deserve separate consideration as these will often take place outside a hospital and can involve a large number of individuals. There is continuing debate regarding which screening methods should be used. It may therefore be worth selecting robust, easy to perform techniques. For example, the use of initial oximetry avoids the discomfort of arterial blood gas sampling and may be justified, particularly in children. The optimal screening intervals are unknown. Current recommendations to screen every 5–10 years,1 ,106 or more frequently if the patient is approaching a period known to be associated with PAVM enlargement and rupture such as puberty2 ,107 or pregnancy,1 ,63 are rarely achieved. Lesions can develop over 2–3 years76 so even these regimes may be insufficiently frequent. The risks of PAVMs increase during pregnancy so it is particularly important to screen prior to or, if necessary, during pregnancy62 ,63 since safe embolisation procedures can be carried out even in the third trimester,62 but it should be noted that desaturation due to right-to-left shunting may be masked by physiological factors during pregnancy.108 Although some authors have suggested that screening for PAVMs can be limited to individuals with particular genotypes,1 the current genetic data do not allow the risk of PAVMs to be confidently excluded and suggest that all HHT families should be offered screening106 ,109 (see below).
TREATMENT OF PAVMS
Surgical resection was the only treatment available for PAVMs until recently and it caused significant morbidity.50 ,71The advent of pulmonary artery embolisation110 ,111altered the risk-benefit ratio of intervention markedly and coincided with a wider recognition of the risks of leaving asymptomatic PAVMs untreated (although such concerns were first raised nearly 50 years ago86). The hazards of intervention relate predominantly to the procedure, though removal of a low resistance shunt may rarely aggravate coincidental pre-existing pulmonary hypertension resulting from nonHHT pulmonary vascular pathogenic events.73 ,76 ,112 ,113
Embolisation
The embolisation techniques used to occlude the feeding vessels to PAVMs with thrombus are described elsewhere.73 ,82 ,114-116 The thrombus organises on thrombogenic fibres associated with carefully positioned metallic coils, or as a result of blood stasis due to an occluding balloon. In one CT scan follow up series 96% of PAVMs regressed including 57% within four weeks of embolisation.88 The coil or balloon needs to be small enough to be sited distally to prevent occlusion of a feeder vessel which also supplies a normal capillary bed, but not too small to risk systemic embolisation through the PAVM82 ,115 or the development of collateral flow between the bronchial artery and distal pulmonary artery resulting in recanalisation of the PAVM.77 As a result, detachable coils and balloons have been developed. The choice of specific agent to initiate thrombus formation is a result of personal preference and experience of the operator. Balloons may be better for more distal placement77 ,115 but they carry the risk of deflation prior to permanent occlusion of the vessel.74 ,76 ,117
Embolisation of PAVMs is generally safe (see table 4) and both safety and efficacy improve with experience75 ,78 ,116 as illustrated by the reduction in episodes of air embolism causing transient angina over recent years. All reports document dramatic improvements in the physiological extent of the shunt (table4).65 Exercise capacity may improve even in patients who had not developed hypoxaemia on exertion.73 Lung volumes are generally preserved and there may be improved forced vital capacity if PAVMs had been acting as space occupying lesions.93 The reduction in Tlco often does not improve,73 ,75 presumably because it reflects the involvement of smaller vessels. Once embolised, the feeding artery to the PAVM usually remains occluded according to most series. A particular result in a recent series with a surprisingly high rate of recanalisation (and unusually half of the new feeder arteries being of bronchial artery supply77) may have been due to technical issues, alternative diagnostic methods, or a different patient population. However, it is well recognised that removal of a low resistance shunt may unmask or provoke the development of new PAVMs or new pulmonary artery feeder vessels to the treated lesion. As a result, a series of treatments several months apart may be required, and it is generally recommended that patients should remain under regular review.75 ,76 ,78
Treatment of PAVM in different series of patients. Series completion dates are given as the year of publication if not specified
Unfortunately, the firm clinical impression of a reduction in the occurrence of cerebral events following embolisation therapy has not yet been supported by adequate numerical data. Between 19% and 60% of patients with PAVMs treated by embolisation have residual shunts as measured directly or suggested by persistent hypoxaemia,75 ,76 ,78 ,89 the figure rising to 73% when contrast echocardiography was used.87 Cerebrovascular complications have occurred in treated patients, including one cerebral abscess in the Hammersmith series,75 and two cerebrovascular accidents occurred amongst the seven patients with persistent PAVMs in the “large PAVM” Baltimore/Yale series (45 patients were initially treated).78 Embolisation of progressively smaller vessels has been adopted to reduce these risks, though technical issues limit the feasibility of embolisation, particularly with smaller vessels and diffuse disease.
Surgery
Surgical procedures have largely been supplanted by the embolisation techniques described above.119 In addition to the perioperative risks, there were concerns regarding loss of functioning lung in patients at risk of recurrent disease in non-resected lobes. Some reports demonstrated improved haemodynamics and oxygenation66 following surgery but physiological studies showed significant residual right-to-left shunts in many patients (table 4).
Surgical intervention may be appropriate in some situations since morbidity has been reduced with improved surgical techniques including the use of video assisted thoracoscopy120 which is helped by the subpleural location of many PAVMs.50 A strong case for surgical intervention by choice was proposed relatively recently74 based on poor embolisation data from a single institution, but the findings in this series are not representative of the results elsewhere.78 Surgical resection might be indicated for patients in whom a persistent right-to-left shunt (and embolic risk) persists following embolisation of all feasible vessels.88 Lung transplantation has been proposed for patients with diffuse disease,54 though for most patients the untreated prognosis is unlikely to justify exposure to transplantation associated morbidity.
Conclusions and perspective
Retrospective series of patients with PAVMs that are generally symptomatic indicate that the risks associated with non-treatment52 ,55 ,59 exceed those of any interventional regime (table 4). The overwhelming benefit of embolisation therapy compared with surgical resection is that it spares functioning lung in patients who are at risk of developing new lesions. Nevertheless, it is important to determine whether embolisation is able to prevent paradoxical emboli as satisfactorily as complete surgical excision in individuals where this would have been feasible. Further long term follow up data are required for all treated patients, probably stratified according to the degree of residual shunt, and distinguishing between individuals with and without HHT (the development of new PAVMs is less likely in the latter group).
Embolisation is currently recommended for all PAVMs with feeding arteries greater than 3 mm in diameter.54 ,76 ,121 This is based on technical issues and the diameters of feeding arteries associated with clinical strokes which in four patients ranged from 2.9 mm to 4.6 mm.121 Some centres routinely treat feeding vessels between 2 mm and 3 mm in diameter.116
While feeding vessels of larger calibre will obviously pose the highest risk of embolic events, it has not been established that smaller vessels pose no risk. Such vessels would include those in angiography negative, shunt positive patients, particularly those detected by contrast echocardiography, and may concern a large proportion of patients with PAVMs as 13 of 31 feeding arteries were less than 3 mm in diameter in one survey of CT scans.96 It will be important to follow the clinical progress of these and equivalent cohorts to assess whether the screening methods are too sensitive for clinical use, or are defining a group of patients in whom particularly rigorous follow up is indicated. At present it seems reasonable to extend the recommendations for prophylactic antibiotic therapy from all patients in whom PAVMs are suspected to include patients in whom PAVMs have been treated, unless careful post-embolisation investigations indicate that the residual shunts have been abolished.
In view of the number of situations in which there is a lack of good longitudinal data on the outcome of patients with HHT, it is hoped that current national and international collaborative efforts will be extended to include such studies with agreed protocols, particularly in the areas highlighted in this section.
Current understandings of mechanisms
The identification of mutations in two genes which encode components of the receptor complexes for ligands of the TGF-β superfamily indicates a role for these growth factors in HHT. In this section we present the currently available data that are directing future research into the understanding of the pathogenesis of this vascular disease. There remain considerable gaps in the mechanistic links between genomic mutations and the generation of the diseased blood vessels.
There are good clinical as well as scientific reasons for pursuing the study of the underlying molecular defects in HHT. Performing mutation analysis of endoglin (HHT1) and ALK-1 (HHT2) on a large number of patients should indicate whether specific mutations are related to particular phenotypes or complications, and further advance our understanding of the structure and function of these proteins and their contribution to the pathology of HHT. A molecular diagnostic test is currently under development and, once such a test is shown to be reliable, it will facilitate the identification of patients with HHT and the classification of families.
MOLECULAR GENETICS OF HHT
Linkage studies first identified a locus for HHT on chromosome 9122 ,123 and suggested that a further gene existed.122 Additional families were used to map a second HHT locus to chromosome 12124 ,125 and it is likely that there is at least one more locus.126 A locus on 3p22 was suggested4 but was subsequently shown not to be the case.125 The mutated genes were identified as endoglin on chromosome 94 and ALK-1 on chromosome 12.5
Endoglin (HHT1)
The gene for endoglin lies on the long arm of chromosome 9 in the interval predicted by linkage analyses. A number of mutations have now been characterised and are summarised in fig3.4 ,109 ,127-130 The mutations include deletions and insertions, missense mutations, and point mutations generating premature stop codons. Additional mutations are predicted in promoter or intronic regions.109 These considerations, and the fact that almost all mutations have been unique to a particular family, highlight the difficulties facing mutational screening programmes.

Summary of endoglin mutations. Intracellular refers to detection when transfected into COS cells. FS = frameshift; UTR = untranslated region; Δ indicates deletion.
Not all mutations result in stable mRNA transcripts.109 ,130 Mutant proteins are rarely detectable and, if expressed at all, exist transiently within the cell and do not reach the cell surface (fig 3 128 and unpublished observations). This leads to a decrease in the level of functional endoglin expressed on peripheral blood activated monocytes and umbilical vein endothelial cells in patients with HHT1.128Quantification of the level of mature endoglin expressed using metabolic labelling and immunoprecipitation is being used to screen potential patients with HHT1 prior to mutation identification (unpublished data, presented in table 5). Affected members in 57 of 95 HHT families tested had a mean endoglin level of 48% of normal (range 8–72%) while non-affected siblings in these families had a mean level of 105% (range 73–140%). The normal levels of endoglin predicted in patients with HHT2 were confirmed in families in which ALK-1 mutations were identified; their levels were indistinguishable from normal individuals. Peripheral blood mononuclear cells (lymphocytes and monocytes) do not express significant levels of endoglin when first isolated but induction occurs if the monocyte fraction is activated by adherence and cell culture for 16–24 hours.128 ,131Levels are, however, 5–20 times lower than on human umbilical vein endothelial cells (HUVEC) where endoglin expression is abundant and constitutive. This explains the smaller range of values observed for HUVEC from patients with HHT1 (26–61%) and HHT2 or normal neonates (83–128%) than for the activated monocytes (table5).
Analysis of endoglin levels in HHT families in relation to mutations detected
These data also indicate that endoglin mutations are not affecting the normal allele, contrary to a previous proposal that the mutations were behaving as dominant negative alleles.127 We have also shown that vessels that appear to be normal in patients with HHT1 express reduced levels of endoglin in situ (50%) compared with values seen in normal individuals and when compared with the endothelial cell marker PECAM-1.132
The next mechanistic question is whether the development of an arteriovenous malformation requires a “second hit” to inactivate the normal copy of endoglin, analogous to the situation proposed for tumour suppressor genes.4 This appears not to be the case as the ratio of endoglin to PECAM-1 was similar in the vascular lesions (cerebral and pulmonary AVMs) as in the other vessels in these patients.132 This suggests that the endoglin mutations are operating as null alleles causing haploinsufficiency.
ALK-1 (HHT2)
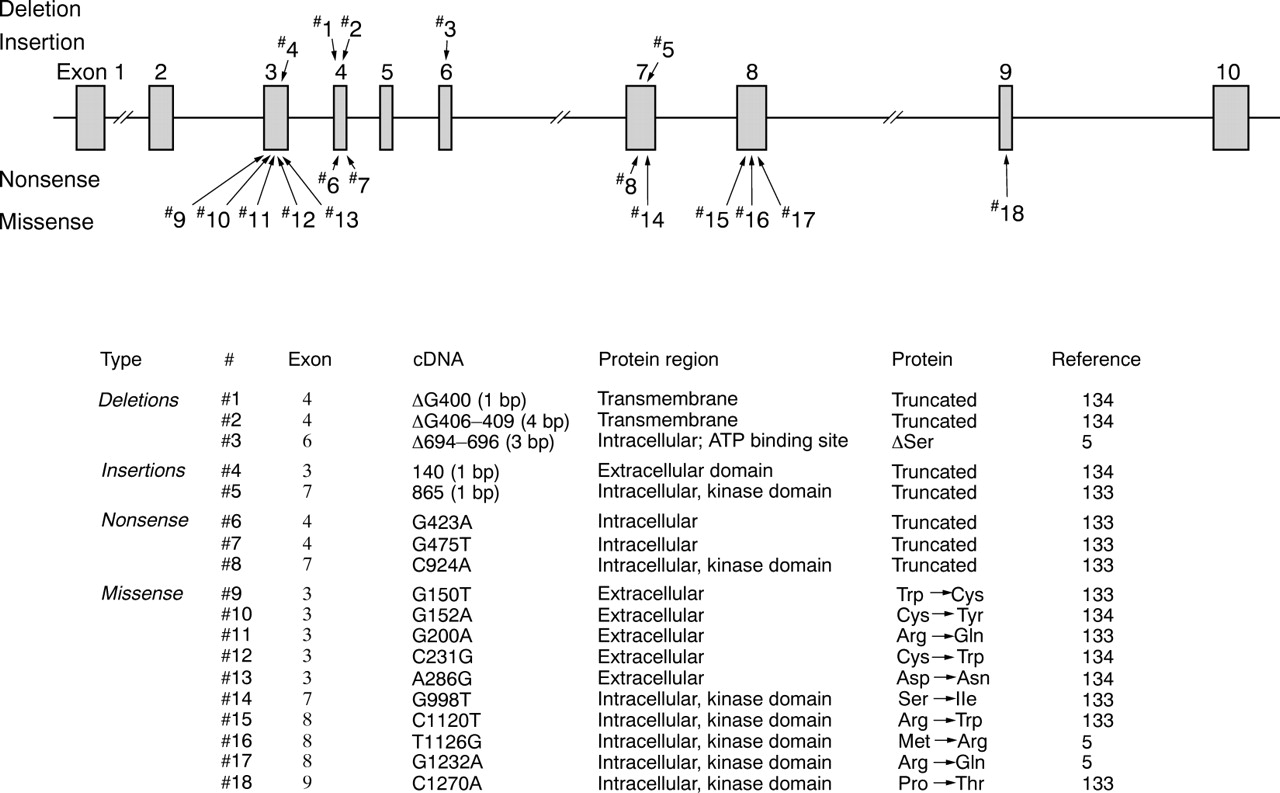
The HHT gene on chromosome 12 encodes a receptor for the TGF-β superfamily, the activin receptor-like kinase 1 (ALK-1; also known as TSR1).5 The mutations described in this gene are found in sequences encoding the extracellular, transmembrane, and kinase domains (fig 4).5 ,133 ,134 Their distribution, and the fact that some mutant alleles appeared to result in low to undetectable levels of transcript, indicate that this group of mutations may also result in functionally null alleles.133

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Overview of ALK-1 mutations.Δ indicates deletion.
GENOTYPE-PHENOTYPE CORRELATIONS
Locus heterogeneity: differences between endoglin and ALK-1 families
Prior to the molecular studies the fact that HHT was a heterogeneous disorder had not been fully appreciated. Occasional familial clustering of cerebral135 ,136 and pulmonary119 involvement had been described but it was not clear whether this represented chance occurrences in a disease in which only some members of an affected family developed any particular complication.
Even before mutations in endoglin on chromosome 9 and ALK-1 on chromosome 12 were described, a flurry of reports signalled that there might be differences between families according to whether they were linked to the chromosome 9 locus or not.137-139 Once mutated genes were identified the search for phenotype-genotype correlations intensified. In essence, however, only two distinctions can be made at present—namely, that PAVMs are significantly more common in endoglin than non-endoglin HHT families1 ,140 and that ALK-1 families tend to have milder disease with more cases of non-penetrance.5 PAVMs do not occur exclusively in endoglin families, however,109 ,122 and the phenotype of endoglin families can be mild for several generations before an affected family member presents with PAVMs.106 ,109 Thus, it is dangerous to apply these population statistics to individual cases.
There are anecdotal suggestions that CAVMs may also be more common in families with endoglin mutations but no evidence has been published. Finally, an HHT family unlinked to eitherendoglin or ALK-1may have a predisposition towards hepatic involvement,126but interpretation needs to be cautious since systematic hepatic screening of asymptomatic family members has not been repeated in other HHT populations.
Specific mutations
Phenotypic data are only available for 30 individuals with seven endoglin mutations (#1, 2, 4, 6, 14, 17, 19, fig 3). No significant differences between mutations or mutational types with respect to age of presentation, severity of nose bleeds, telangiectasia, or PAVMs have been identified.109 This would be predicted from a haploinsufficiency model. Each mutation accounted for a spectrum of disease severity in affected family members not entirely accounted for by age-related penetrance,109 highlighting the importance of additional modifiers of the HHT phenotype.
Genetic and environmental modifiers of the HHT phenotype
Clinical observations provide clues to several factors that may potentially influence the development of HHT lesions in patients, though undoubtedly more will be described. Defects in the coagulation cascade which could exacerbate a haemorrhagic tendency are often cited and are discussed in detail elsewhere,141 but they are rare and have not influenced clinical practice. Variations in fibrinolysis may also contribute to a haemorrhagic tendency: quenching excessive fibrinolysis using aminocaproic acid had beneficial effects in some23 ,24 but not all25 of a handful of patients tested.
Less obvious at first sight is the importance of the patient’s sex in a condition inherited as an autosomal dominant trait. Excluding neonates, women are more at risk of developing PAVMs52 ,53 ,55 ,63 ,75 ,76 and possibly hepatic involvement33 ,142 and cerebral haemorrhage.143 This may reflect a fundamental modification of the HHT vasculature by female hormones, or relate to haemodynamic changes during pregnancy.63 A role for direct hormonal influences is supported by the successful treatment of HHT related gastrointestinal bleeding using combined oestrogen-progesterone therapy,21 a report of progesterone receptors in vessels of patients with HHT,144 and variations in epistaxis during the menstrual cycle and menopause.63 Haemodynamic changes can also exacerbate PAVMs, including the development of pulmonary hypertension as a result of mitral stenosis145or left ventricular dysfunction.146 Furthermore, cutaneous telangiectasia (known to be in a dynamic state in HHT147) have been observed to regress following successful pneumonectomy for PAVMs,80 ,148 possibly due to a predicted fall in cardiac output though this was not documented.
MECHANICS: HOW TO GENERATE AN HHT VESSEL—THE EXAMPLE OF PAVMS
The question as to whether any of the abnormal blood vessels in patients with HHT (telangiectases or larger arteriovenous malformations) represent true congenital malformations rather than an acquired lesion in intrinsically abnormal vessels has not been resolved. The majority of PAVMs present during teenage and adult life, suggesting that vascular remodelling is occurring. However, the presentation of PAVMs in childhood is well recognised50 ,52 ,53 ,55 ,58 ,59 ,71 ,75 ,78 and, although severe disease in two infants may have been due to homozygous HHT, the possibility of PAVMs arising during development cannot be ruled out.
As vascular lesions are associated with both HHT1 and HHT2, the potential role of both endoglin and ALK-1 needs to be considered. Both mutated genes encode proteins that are expressed predominantly in endothelial cells, which might explain why affected individuals are generally well apart from their vascular pathology. As there is only limited information available on ALK-1 we will discuss it first before concentrating on endoglin and the lesions with which it is particularly associated—namely, PAVMs.
ALK-1: a serine/threonine kinase type I receptor in search of a function
Members of the TGF-β superfamily bind and signal through a heteromeric receptor complex composed of serine/threonine kinases in which the type II receptor generally binds ligand and phosphorylates the type I receptor which in turn signals through the recently identified cascade of Smad proteins.149 ,150 ALK-1 is a serine/threonine kinase type I receptor that can associate with either TGF-β-RII or activin-RII when co-transfected into COS cells (a transformed primate cell line), and can bind TGF-β1 or activin, respectively, although with low affinity.151-153 Since this has not been demonstrated with endogenous receptors, ALK-1 is referred to as an orphan receptor as its physiological ligand has yet to be identified. The potential ligand might belong to another subgroup of the TGF-β superfamily—namely, the bone morphogenetic proteins (BMPs), as activation of ALK-1 has recently been shown to trigger an intracellular Smad-1 pathway associated with signalling by BMPs.154
High levels of ALK-1 are found on human endothelial cells and in lung and placenta, both of which are highly vascular.151 ,152Rat ALK-1 was found to be most abundant in pulmonary blood vessel endothelium (all types) as well as on aorta, vena cava, and certain blood vessels of kidney, spleen, heart, and intestine.155Lung expression increased fivefold in the early postnatal period, most probably reflecting an increase in the pulmonary vasculature.155 ALK-1 was also found on rat splenic macrophages and a murine bone marrow stromal cell line.156This distribution has many parallels to that of endoglin. A more recent study in a murine model found that the distribution of ALK-1 was highest around eight days after conception at sites of vasculogenesis in both embryonic and extra-embryonic tissues, in giant trophoblast cells, and in the endothelial lining of blood vessels in the decidua. From days 9–12 ALK-1 was highest in blood vessels, lung mesenchyme, the submucosa of the intestine and stomach, and at sites of epithelial-mesenchymal interactions.157 This pattern of expression of ALK-1 is very close to that of TGF-β1,158in keeping with a potential role in vascular development which may be confirmed in the ALK-1 null mice currently in development.
Endoglin: an accessory protein of the TGF-β receptor superfamily
Endoglin was first identified in childhood leukaemia with a pre-B lymphocytic phenotype159 and was soon recognised as an endothelial cell marker (CD105) which is expressed on all types of vascular endothelium.160 ,161 It is also present on bone marrow mononuclear cells of the pre-erythroblast lineage162 and on activated macrophages.131The expression of endoglin is transiently increased in mesenchymal cells during embryonic development in association with cardiac valve formation, for example.163 Endoglin expression is also increased in mesenchymal cells in the human lung starting at eight weeks gestation and remains high at 20 weeks (unpublished observations), in keeping with a potential role in vascularisation of lining mesenchyme. In the adult endoglin is expressed on endothelium160 and placenta,164 with smooth muscle cell expression also described.165 Endoglin is upregulated on vascular endothelial cells in tumours,166pathological skin lesions including psoriasis, and in response to UV irradiation.167 ,168
Endoglin is a homodimeric membrane glycoprotein of apparent Mr = 180 000169 ,170 and has been shown first by chemical crosslinking in HUVECs and subsequently in fibroblasts to bind TGF-β1 and TGF-β3 but not TGF-β2.170-172 It can associate with TGFβ-RII and TGFβ-RI (ALK-5)173 ,174 as indicated by immunoprecipitation of the TGF-β1 affinity crosslinked complexes with antibodies to either endoglin or TGFβ-RII. However, we have recently demonstrated that endoglin by itself does not bind TGF-β1/β3 and therefore cannot be referred to as a receptor; it only behaves as such when associated with TGFβ-RII.175
We have also observed that endoglin can bind other growth factors of the TGF-β superfamily when associated with the different ligand binding receptors. For example, endoglin can bind activin or BMP-7 when co-transfected with activin RII; it will not bind BMP-7 via BMP-RII, however, indicating that these interactions are specific. It will also bind BMP-2 when associated with the ligand binding type I receptors, ALK-3 and ALK-6.175 These results suggest that endoglin might serve as an accessory protein for multiple kinase receptor complexes of the TGF-β superfamily and that it could perform a different regulatory role in diverse cell types, according to the kinase receptors, ligands, and Smad mediators present.
When transfected into U937 monocytes or rat myoblasts, endoglin can modify certain TGF-β1 (but not TGF-β2) responses such as the inhibition of cell proliferation by downregulation ofc-myc, the increase in homotypic adhesion mediated by increased fibronectin production, and modulation of the fibrinolytic system by synthesis of plasminogen activator inhibitor-1.176 ,177 Conversely, antibodies and antisense oligonucleotides to endoglin added to first trimester trophoblast explants in culture stimulated the differentiation of trophoblasts into invasive cells, a process necessary for establishing fetal-maternal interactions and known to be inhibited by TGF-β1/β3.178
Although a potential role for endoglin in regulating responses to ligands other than TGF-β1 must be kept in mind while trying to elucidate the underlying mechanisms of HHT, it is clear that TGF-β1 is an important regulator of vascular development. TGF-β1 null mice showed a primary defect in yolk sac vasculogenesis and haematopoiesis which led to death at around day 10 in 50% of homozygous and 25% of heterozygous mice.179 The differentiation of endothelial cells was affected (rather than their initial appearance or outgrowth from yolk sac mesoderm), causing inadequate capillary formation and weak vessels with reduced cellular adhesiveness. Furthermore, TGF-β3 null mice died soon after birth with extensive pleural haemorrhage and dilated and fragile pulmonary veins and capillaries.180 ,181 This suggests that TGF-β1 and TGF-β3 are both implicated in the development of lung vasculature and impairment of their usual function might contribute to the pathology of PAVMs.
With this information is it possible to propose why PAVMs develop? Endoglin is present from the earliest stages of pulmonary vascular development, and TGF-β1 and TGF-β3 are implicated in vasculogenesis180 ,181 and angiogenesis,182both processes being critical in vessel development and maturation.183 In patients with HHT and reduced endoglin expression, vascular development is sufficiently normal for most individuals to have apparently normal pulmonary vasculature. The careful regulation of endoglin expression (and ALK-1) during development suggests, however, that their complete absence could be lethal, as seen in the potential homozygotes.12 ,13Current studies in null mice should resolve this issue.
If most vessels with 50% expression of endoglin develop normally, what additional factors cause some vessels to develop into PAVMs? Certain physiological or pathological conditions, including altered blood flow184-186 and hormonal changes, could be important. This would provide a partial explanation for why only a small proportion of endothelial cells expressing a mutant allele are involved in morphologically abnormal HHT vessels.
We would like to propose that reduced levels of functional endoglin results in blood vessels that are more susceptible to dilatation and remodelling. We have shown that the endothelium in a CAVM was partially disrupted and stretched out so that the density of important surface molecules was reduced.132 In addition, it may be recalled that TGF-β1 plays a major role in wound repair. It is abundantly released by platelets and macrophages at sites of inflammation and injury,187-189 is upregulated by shear stress,190 and has been implicated in vascular repair processes.191 In tissue repair as well as in development, TGF-β1 stimulates the growth of cells of mesenchymal origin. Furthermore, TGF-β1 induces the synthesis of extracellular matrix proteins, their integrin receptors on the cell surface, and the protease inhibitors implicated in their degradation such as plasminogen activator inhibitor and tissue inhibitor of metalloproteases; it also downregulates the expression of matrix degrading enzymes such as collagenase.192 ,193 The net effect of TGF-β1 is thus to stimulate matrix production and enhance interactions between cells and matrix and between endothelium, smooth muscle cells, and mesenchymal cells in the vessel wall. Vasodilatation, intravascular pressure, or shear stress can affect endothelial cell shape in vivo and could initiate remodelling in vessels with reduced levels of endoglin.
Perspectives
The abnormal vascular structures in HHT appear to develop because mutations in endoglin, ALK-1, and possibly other genes result in a dysregulated response to ligands of the TGF-β superfamily which play complex and important roles in vascular development and repair. At present we cannot pinpoint the exact ligand(s) or manner in which normal vascular development or homeostasis is perturbed by these mutations. Potential ligands of the TGF-β superfamily have diverse and often multiple effects that may influence the vasculature, effects which may be differentially regulated and fine tuned according to the exact amount of endoglin or ALK-1 present. Future developments are likely to include the identification of the precise TGF-β family members involved in the disease, and of additional components of the pathogenic pathway which may be identified from finding mutations in new genes in other HHT families. In addition, clarification of the roles of normal and mutated endoglin and ALK-1 through cellular and animal models is expected.
The identification of a gene defect in an inherited disease leads to expectations of molecular therapies, particularly when the molecular mechanism appears to be stoichiometric insufficiency, amenable to replacement therapy. However, there are hazards in attempting to restore functional levels of a deficient protein in complex regulatory networks such as those in which ligands of the TGF-β family are involved. It may be some time therefore before molecular manipulations of HHT can be applied to patients and, for the foreseeable future, conventional therapies are likely to be required.
Acknowledgments
The cases illustrated in fig 2B and fig 3 were investigated at the Royal Infirmary of Edinburgh. The authors thank Dr David Lamb and Professor Bill MacNee for fig 2B. For fig 3, Dr Kieran McBride performed the angiography, Dr Patricia Tweedale the shunt measurements, and Dr Micheal Sudlow gave permission to report the case. The authors are grateful to Dr Patricia Tweedale, Dr Kees Westermann, and Dr Bob White for helpful discussions, and to Dr Gillian Wallace and Dr Patricia Tweedale for manuscript review. They also thank the staff of the Erskine Medical Library, University of Edinburgh and colleagues who provided them with manuscripts prior to their publication.
Most countries have self-help groups for patients with HHT:
UK: Telangiectasia Self-Help Group, 39 Sunny Croft, Downley, High Wycombe, Bucks. HP13 5UQ, UK.
USA: HHT Foundation International, PO Box 8087, New Haven, CT 06530, USA.
References
Footnotes
Claire Shovlin has been supported by a Wellcome Trust Advanced Fellowship; Michelle Letarte is a Tery Fox Research Scientist of the National Institute of Canada, and is supported by research grants from the Heart and Stroke Foundation and the Medical Research Council of Canada.