Article Text

Statistics from Altmetric.com
In allergic asthma, allergen specific T lymphocytes of the CD4 subset (T helper or Th cells) control the cellular and molecular events underlying the establishment and chronic maintenance of airway inflammation that characterises the disease. The basic immunological event is the shift of allergen reactive CD4 Th lymphocytes towards a Th2 phenotype with the production of Th2 cytokines such as interleukin (IL)-4, IL-13, and IL-5. Th2 cells can support both allergen specific IgE production and eosinophil recruitment.
Recognition of allergen via the T cell receptor (TcR) provides the trigger for a specific T cell response; as any antigen, allergen is seen by Th cells as one of a few possible peptides derived from proteolytic processing. Immunogenic peptides are presented on major histocompatibility complex (MHC) molecules expressed on the membrane of antigen presenting cells. However, antigen recognition can follow such opposite events as anergy or activation of T cells—that is, induction of tolerance versus establishment of an effective immune response. Current concepts recognise that the outcome of antigen recognition is dependent, at least in part, on the complex interaction between MHC molecule associated membrane proteins (collectively known as co-stimulatory molecules) on the antigen presenting cells and their ligands on the T cells. Moreover, when the immune response is activated, co-stimulation is also involved in determining which effector profile will be acquired by the T cells—that is, a Th1 or a Th2 phenotype.
We will briefly review the experimental evidence supporting the concept that interference with the proper co-stimulatory molecules may allow the design of pharmaceutical products which cause immune deviation that could quantitatively and qualitatively change the T cell response. This approach could interfere with the chronic amplification of allergic inflammation at the level of the target organ in atopic individuals.
Co-stimulatory molecules and antigen presentation
Alveolar macrophages and dendritic cells are resident phagocytes in the lung. Physiologically, alveolar macrophages act as very poor antigen presenting cells and, indeed, they are considered as inhibitors of local T cell activation.1 This characteristic of alveolar macrophages is thought to prevent the inappropriate activation of the lung immune system, which is continuously exposed to inert airborne antigens, and thus prevent a persistent inflammatory reaction which could lead to tissue damage and impairment of gas exchange.2
The poor antigen presenting ability of alveolar macrophages in healthy individuals may be related to reduced expression and/or function of membrane co-stimulatory molecules on the cell surface.3 On the other hand, the increased antigen presenting function of alveolar macrophages found in chronic inflammatory disorders such as allergic asthma and pulmonary sarcoidosis4 may be a result of the enhanced expression of co-stimulatory molecules by these cells. Indeed, in different murine models of asthma the key role of the expression of two co-stimulatory molecules (CD80 (or B7.1) and CD86 (B7.2)) on alveolar macrophages in the maintenance of allergic inflammation has been progressively elucidated.
In contrast to alveolar macrophages, dendritic cells constitutively express co-stimulatory molecules such as the B7 molecules CD80 and CD86. Dendritic cells are located in the interstitial lung tissue5 and act as highly efficient antigen presenting cells carrying inhaled particles with antigenic properties (usually from pathogens) to the regional lymph nodes.6 In these organised immune sites naive T cells bearing the relevant specificities are located. In response to antigen presentation, allergen specific T and B cells are expanded. In this fashion they acquire the phenotype of “memory” lymphocytes with recirculation capabilities (dependent on a defined array of homing receptors7) allowing them to reach the lung epithelium and activate their effector function at this site.
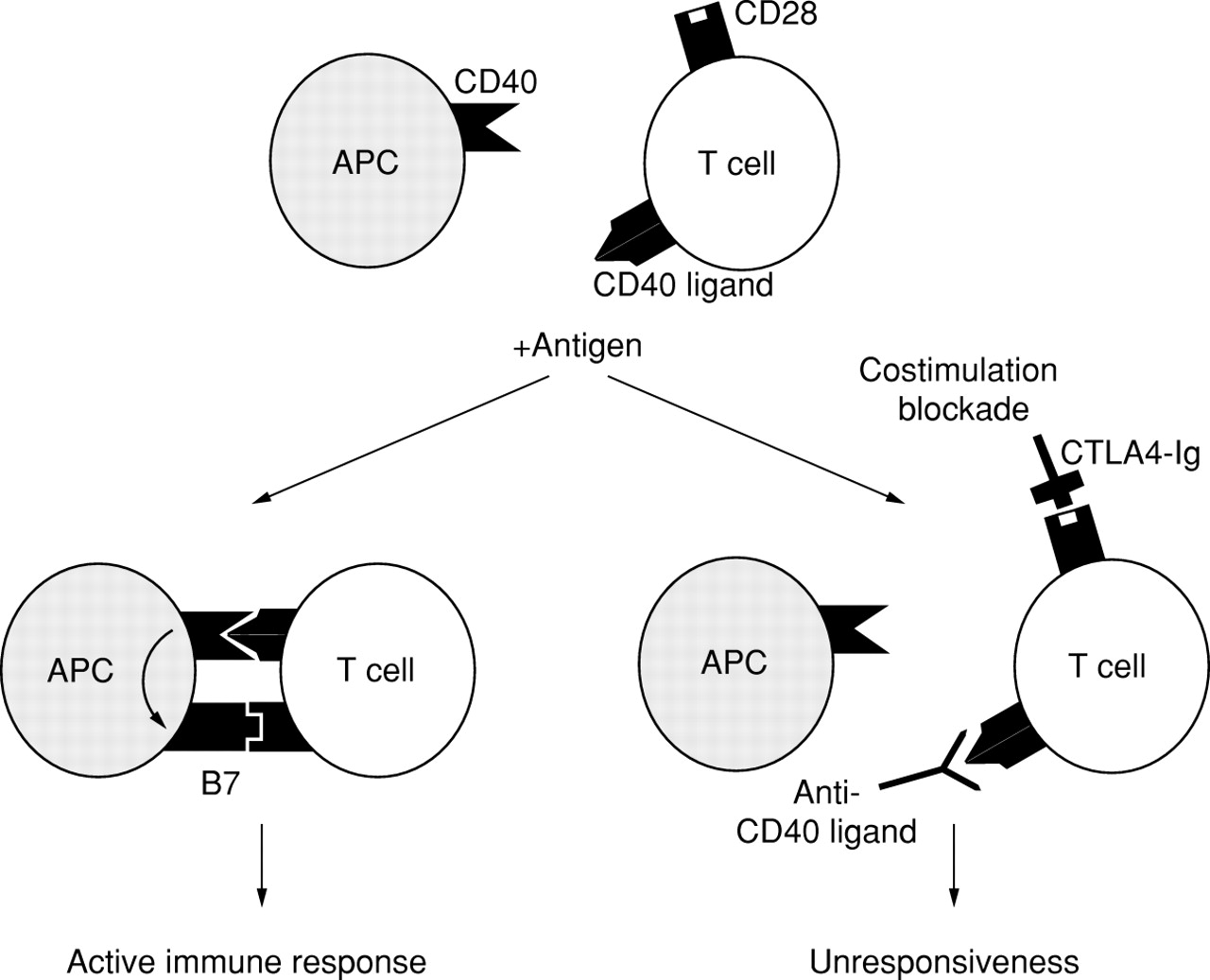
The presence of CD80 and CD86 molecules on antigen presenting cells provides a major co-stimulatory signal for activation of T cells through the T cell counter receptor, the CD28 molecule (fig1).8 Antigen recognition, in the absence of CD28 co-stimulation, results in lack of IL-2 mRNA induction9and in T cell anergy.10 In contrast, in the presence of CD28 co-stimulation, antigen recognition results in IL-2 mRNA stabilisation and accumulation and in T cell activation.11

{kind=link}
Antigen presenting cells express the CD40 molecule (alveolar macrophages from patients with sarcoidosis express this molecule while healthy controls do not). The corresponding ligand (CD40 ligand or CD153) is constitutively expressed on lymphocytes, but its expression is upregulated on encounter with an antigen. These events further upregulate expression of B7 molecules on antigen presenting cells, thus allowing the engagement of the CD28 molecule on T cells. At this point, an effective antigen specific immune response can develop. Molecules which interfere with one or both co-stimulatory pathways may be promising candidates as innovative anti-inflammatory drugs.
Upon activation an additional ligand of CD80 and CD86, the CTLA-4 molecule, is expressed by T cells.12 ,13 CTLA-4 binds the B7 molecules CD80 and CD86 with 20–50 times higher affinity than does CD28 but, in contrast to CD28, delivers downregulatory signals (table1).13
Co-stimulatory molecules and their ligands
The B7 molecules are expressed by antigen presenting cells in a fashion which depends on the type and on the state of activation of the cells. Indeed, blood monocytes constitutively express CD86, while CD80 is induced only upon activation of these cells. In contrast, dendritic cells, which are more potent antigen presenting cells, constitutively express both CD80 and CD86.
Antigen presentation by non-professional antigen presenting cells on normal healthy tissues are, in principle, either tolerance inducing or do not support the maturation of the immune response because of the absence of co-stimulatory molecules. Indeed, while the expression of MHC molecules on activated endothelium14 ,15 and epithelium16 ,17 is regulated by several stimuli, usually this is not associated with the expression of B7 co-stimulatory molecules. However, in some circumstances other membrane molecules can serve this function—for example, LFA-318 or ICAM-1.19 Notably, human Th1 and Th2 clones were reported not to be susceptible to inhibition by CD80 or CD86.20Moreover, Yi-qun et al reported that, in vitro, activated T cells could be restimulated in the absence of CD80 or CD86 co-stimulation.21 It may therefore be possible that the expression of class II MHC molecules in vivo by non-professional antigen presenting cells could support the proliferation of defined lymphocyte subsets in peripheral tissues. It is likely that these complex interactions differentially contribute to the establishment and amplification of several inflammatory reactions in single target organs, depending on the phenotype of the T cells on the one hand and the type and state of activation of antigen presenting cells on the other.
The expression of the B7 molecules may also be upregulated by the interaction between other co-stimulatory molecules expressed by antigen presenting cells and T cells such as the CD40-CD40 ligand or CD154.22 This pathway, initially described as having a role in B cell activation, has been recognised as having a key role for T cell activation also.22 CD40 is expressed on antigen presenting cells such as macrophages, dendritic cells, and B cells, as well as by other cell types such as endothelial cells.23The ligand for CD40 not only signals positively for antibody production by B cells, but also induces strongly the expression of B7 molecules24 ,25 and the secretion of inflammatory cytokines that participate in T cell activation.26
Lessons on the role of CD80 and CD86 in immunity and tolerance from knock out mice
Mice genetically deficient in CD80 or CD86 and double knock out mice have recently been generated.27 ,28 Mice lacking either CD80 or CD86 remained capable of generating T cell dependent humoral immune responses, suggesting that the co-stimulatory functions of these molecules are redundant. However, in the same studies CD86 knock out mice were less responsive to immunisation than CD80 knock out mice, suggesting a primary role for CD86 in the initiation of immune responses. Furthermore, both Th1 and Th2 responses were reduced in CD86 knock out mice, indicating that immunodepression is not associated with immunodeviation. Finally, CD80/CD86 double knock out mice showed the most severe impairment in their immunity, supporting the view that CD80 and CD86 act cooperatively to modulate T cell activation. Recently, Schweitzer et al reported that induction of IL-4 production and Th2 differentiation by naive T cells is highly dependent on B7 molecules, whereas IL-4 production by previously activated T cells is not.29 These authors found that the predominant contribution of B7 mediated signals to Th1 cytokine production by both naive and primed T cells was on IL-2 production (and expansion) rather than on IFN-γ (effector cytokine) production. They therefore concluded that the antigenic experience of a T cell at the time of B7 blockade may determine whether blockade predominantly affects T cell expansion, differentiation, or effector cytokine production. These results indicate the variables that need special attention in evaluating the effect of co-stimulation blockade in any given experimental system. It will be interesting to evaluate the characteristics of experimentally induced asthma in B7 knock out mice to explore different functional roles for CD80 and CD86 in these models (see below).
Involvement of CD80 and CD86 in allergic asthma
The CD80 co-stimulatory molecule is abnormally expressed on alveolar macrophages, both at baseline and after allergen challenge, in atopic individuals with asthma.30 ,31 There were no differences in the alveolar macrophage expression of CD86 between healthy controls and allergic asthmatic subjects, both at baseline and after allergen challenge. In in vitro experiments to characterise the functional relevance of this finding, we observed that the secretion of IL-4 and IL-5 by autologous allergen specific Th2 cells, as well as their in vitro proliferative response to allergen, were all highly dependent on CD80.32 Interestingly, in allergic asthma the proportion of alveolar macrophages expressing CD80 was highly correlated with eosinophil recruitment to the lung induced by allergen inhalation challenge, further stressing the relevance of this co-stimulatory molecule in the maintenance of inflammation in the allergic lung. It is likely that the presence of the molecular prerequisites for an efficient antigen presenting function can explain the expansion of allergen specific Th2 CD4 cells in the lower respiratory tract of allergic asthmatic subjects32 but not in non-atopic controls.33
Larché et al 34 have recently reported that co-stimulation through CD86 is involved in the expansion of T cell responses to allergen in atopic asthmatic subjects; they found that both allergen induced proliferation and IL-5 secretion were inhibited by CTLA4-Ig fusion protein and anti-CD86 (but not by anti-CD80) monoclonal antibodies. In particular, they reported that IL-5 production is inhibited by CD86 blockade in a manner independent of IL-2 since exogenously added IL-2 could restore T cell proliferation but not IL-5 production. Moreover, they found that T cells from bronchoalveolar lavage fluid can proliferate on addition of allergen in a CD86 dependent fashion. However, these authors could not formally conclude that CD80 contributed to the activation of allergen specific T cells in asthmatic subjects in defined steps of initiation, maintenance, and amplification of the allergic immune response. Although the mechanisms of local amplification of the immune response may be additional to the recruitment of lymphocytes from peripheral blood,35 ,36 these results suggest that co-stimulation treatment targeted at T cells could be of considerable interest in allergic asthma.
Therapeutic potential of co-stimulation blockade
A recombinant fusion protein consisting of the extracellular domain of CTLA-4 linked to the constant region of IgG1, known as CTLA4-Ig, binds B7 molecules with an affinity similar to membrane CTLA-4. It therefore acts as a powerful inhibitor of B7/CD28 mediated co-stimulation (fig 1).37 Interestingly, in murine models of allergic asthma CTLA4-Ig had a powerful inhibitory effect on the allergic inflammation and its functional consequences.38 ,39 This protein could therefore represent a powerful “immunomodulator” with the potential to suppress an unwanted ongoing immune response to allergens in humans.
Lack of systemic effect is expected since this molecule has also been produced as a chimera consisting of the human IgG constant region engineered with the extracytoplasmic domain of human CTLA-4. Moreover, CTLA4-Ig did not produce significant side effects in large animal models when administered systemically.40 It is tempting to speculate that its use by inhalation could offer further advantages in terms of flexibility, incidence of side effects, and cost.
A further possible method of interfering with co-stimulation is offered by CD40 ligand antagonists. Blocking the CD40/CD40 ligand co-stimulatory pathway by means of a monoclonal antibody against murine CD40 is effective in preventing allograft rejection in mice.41 It remains to be tested in animal models whether this approach may be relevant in controlling the allergic immune response. Moreover, the combined administration of CTLA4-Ig and anti-CD40 ligand could be even more effective, as suggested by recent reports in renal allograft models in mice41 and in non-human primates.42
It is still controversial whether, and to what extent, CD80 and CD86 provide qualitatively different signals to T lymphocytes which are progressing towards a given differentiation pathway (Th1 or Th2). For instance, CD80 and CD86 have distinct regulatory functions in animal models of Th1 autoimmune diseases.43-45 In murine models of allergy and asthma, independently set up by different groups, distinct roles have been attributed to CD80 and CD86 with regard to formal gain and loss of function.46-48 The results have been contradictory; Keane-Myers and Harris reported a dependence on CD86 in the establishment of allergic inflammation while Tsuyuki and collaborators concluded that CD80 was primarily involved in their animal model.
Attempts to obtain CD80 and CD86 specific ligands have been successfully carried out. Morton et alreported that five single amino acid substitutions within the CTLA-4 MYPPPY domain, while exerting a modest effect on CD80, completely abrogated CD86 binding.49 Moreover, substitutions in the N-terminal of the MYPPPY region and within the CDR1-like region of CTLA-4 eliminated both CD80 and CD86 binding. Hence, CD80 and CD86 have distinct binding sites on CTLA-4.47
Harris et al explored the effect of a mutant version of CTLA4-Ig in a mouse model of allergy and asthma and found that CD80, but not CD86, was necessary for the maintenance or amplification of lung inflammatory responses.47 Thus, the availability of molecules capable of interfering selectively with key co-stimulatory receptors on the membrane of alveolar macrophages might represent a relevant improvement in the search for refined immunomodulators.
Conclusions
In allergic asthma the enhanced co-stimulatory activity exerted by alveolar macrophages to resident lung T lymphocytes is probably involved in the establishment and maintenance of chronic inflammation in the lower respiratory tract. Specific molecular tools capable of interfering with this upregulated function might allow the design of a new category of anti-inflammatory drugs in the fight against the immunological basis of inflammation in asthma.
Acknowledgments
This work was supported by a grant from Istituto Superiore di Sanità to SEB.