Article Text

Statistics from Altmetric.com
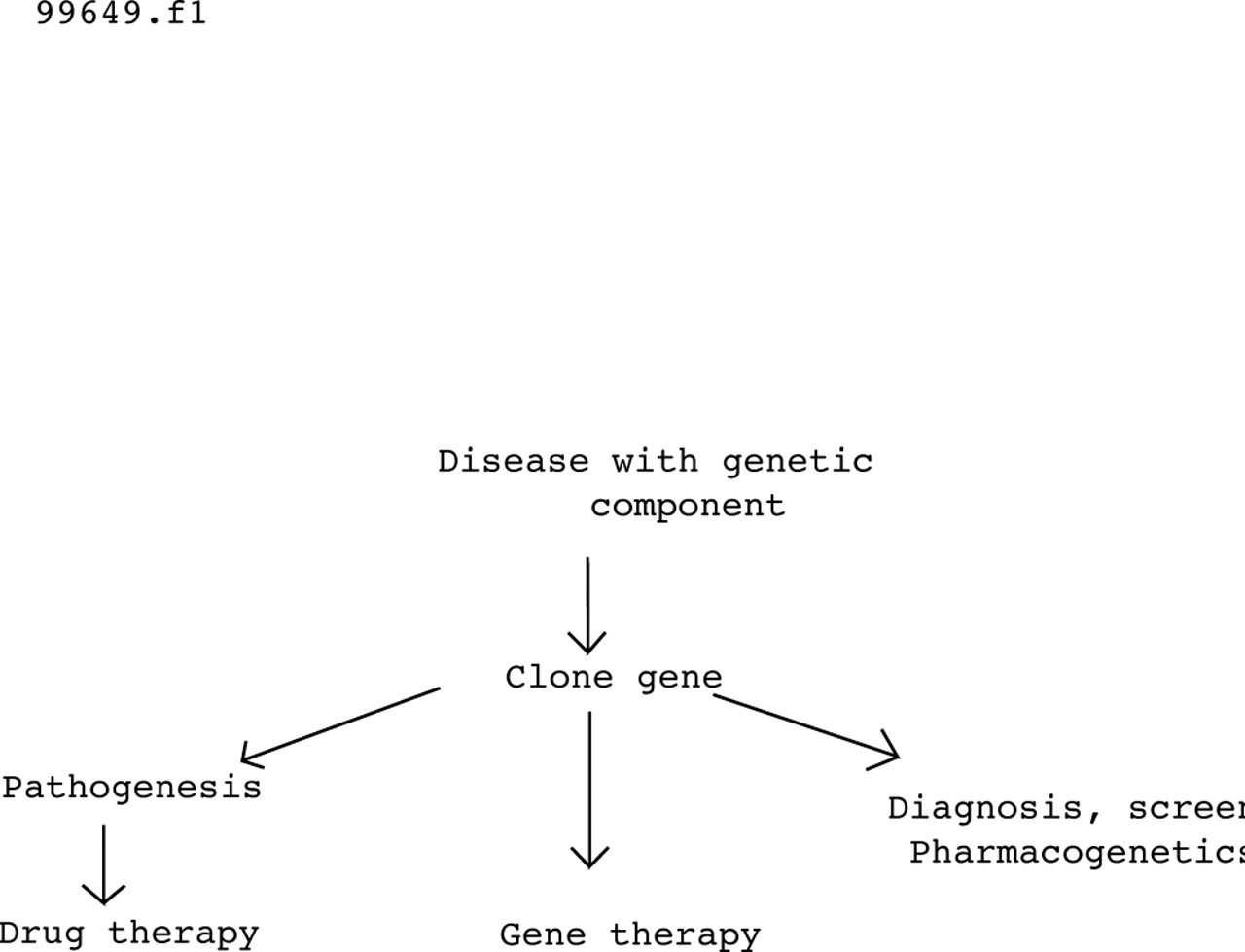
These days everyone is looking for genes. Grant money, media interviews, self-esteem, Nobel prizes, and lots of interesting biology lure researchers into molecular genetics, and diseases as diverse as asthma, chronic obstructive pulmonary disease and lung cancer will soon be mastered by brave new understanding. All true, but will it make any difference? Cystic fibrosis got there first, and 10 years after the discovery of the CF gene it is a good time to take stock. Compared with polygenic diseases, cystic fibrosis is easy and therefore paints the rosiest picture of what can be achieved by genetic research. If the CF gene has helped patients, then the discovery of genes associated with other diseases may do the same. The steps involved in the genetic revolution were well illustrated in a recent review article1 as shown in fig 1.
{kind=link}
Medical consequences of the human genome project. Reproduced with permission from Collins.1
Pathogenesis
By sequencing the CF gene, the protein was deduced and found to resemble a family of ATP binding membrane proteins with both ion channel and macromolecule transporter properties. Cystic fibrosis transmembrane conductance regulator protein (CFTR) is a chloride channel which opens in response to phosphorylation by ATP and is found in sweat and pancreatic ducts, gut, seminiferous tubules, and conducting airways—all sites of cystic fibrosis disease.2Interestingly, CFTR is also expressed in the heart, choroid plexus, and renal tubules, organs with normal function in cystic fibrosis. Other properties of cystic fibrosis have been proposed such as the regulation of apical membrane sodium transport in the airways and acidification of intracellular organelles. No transporter function of CFTR has yet been found.
Over 800 disease associated mutations of the CF gene have been described; these differ with ethnic origin but only a handful are common. These mutations have been classified according to their molecular pathology into five groups: (1) no protein, (2) defective intracellular trafficking (typically the commonest mutation ΔF508), (3) and (4) altered channel function, and (5) reduced levels of functional protein, and this mechanistic information may point the way to new pharmacological treatments.3 The link between the various mutations and disease is still being worked out; abnormal channel function goes with altered electrolyte and water content of secretions, thus explaining high sweat electrolytes and dehydrated viscid pancreatic secretions. The pathogenesis of lung disease is, however, more controversial and there are three schools of thought, all of which concern a defect in bacterial defences. The simplest is based on the finding of increased bacterial adherence to respiratory epithelial cells due to altered cell surface glycoproteins.4 (Defective ingestion of bacteria by epithelial cells has also been proposed.) The second and third are based on airway surface liquid changes. A high salt school proposes that increased sodium and chloride levels limit the activity of antibacterial defences such as lysozyme and defensins, while a low volume school proposes that increased sodium and consequently water absorption from the airways leaves a depleted sol layer which impairs mucociliary clearance. All three mechanisms may co-exist and each is open to pharmacological correction. Finally, one of the surprises of cystic fibrosis research is how poorly genotype predicts phenotype and, although the typical presentation of cystic fibrosis with pancreatic insufficiency goes with ΔF508, the high variability in clinical expression of lung disease is not yet explained.
From pathogenesis to treatment
This increased understanding has led to the investigation of a range of possible new treatments. Firstly, knowledge of the molecular pathology has suggested various agents to increase gene expression (phenylbutyrate, gentamicin), enhance trafficking (glycerol), or enhance ion channel activity (milrinone, CPX). Naturally, such approaches will only work with the appropriate genotype. Phenylbutyrate and glycerol have shown promise in initial clinical trials but greater potency and precision of action are still needed. Secondly, an understanding of organ pathophysiology has suggested ion transport modifying drugs, although admittedly this line of investigation was being pursued before the discovery of the gene. Agents include amiloride5 or benzamil to reduce sodium absorption6 and UTP to enhance chloride secretion, all of which are at different stages of testing in human trials. These approaches are logical to either high salt or low volume theorists. In addition, those in favour of the high salt theory would favour some method of adding water to airway surface liquid (ASL) or developing salt resistant defences while proponents of the low volume theory favour addition of salt and water to ASL, perhaps by nebulising hypertonic saline or some other less well absorbed osmolyte. Finally, the sheer number of possible drug mechanisms which may be involved has persuaded others to screen the whole pharmacopoeia in a mindless (but intelligent) medium throughput screening system to see if any agent already in use might work in cystic fibrosis.
Diagnosis and screening
Before the discovery of the gene some were predicting a single mutation which would greatly simplify both diagnosis of cystic fibrosis and carrier detection, as well as opening the door to population screening and eventual eradication of the disease. Instead, the discovery has, if anything, made things more difficult. The large number of mutations together with the finding of new gene associated disorders has made diagnosis more complex. The typical patient with cystic fibrosis usually has one of the four commonest mutations so genotype analysis confirms the diagnosis in those for whom no confirmation is really needed. For patients with borderline clinical disease, genotyping may reveal one or none of the common mutations and, since routine laboratories cannot take on the task of screening for all 800 + mutations, the situation usually remains unclear. The presence of two disease associated mutations is diagnostic, the finding of one may be suggestive but is certainly not diagnostic since the carrier frequency in the population is high (1:25 in the UK), and the failure to find a mutation in no way excludes the disease. Furthermore, studies have now found convincing linkage between some mutations of the CF gene and idiopathic pancreatitis or male infertility in people with no other evidence of cystic fibrosis, so extending the clinical expression of the disease. Preliminary reports have also suggested linkage to allergic bronchopulmonary aspergillosis, while mutations of the CF gene in association with asthma have been reported as more than expected, less than expected, and just about right. So, while genotyping sometimes makes the diagnosis of cystic fibrosis more certain, the exclusion of cystic fibrosis and knowing precisely what a diagnosis of cystic fibrosis means has become more difficult.
In contrast, genotyping has greatly assisted the identification of carriers of the CF gene and provides clear cut benefits. Such screening has not as yet been applied to healthy populations as a whole but has been undertaken in people with a known risk of cystic fibrosis and in antenatal clinics. When there is a known risk of cystic fibrosis—for example, in an affected family—genotyping provides reassurance when no mutation is found or allows better informed reproductive decisions when the test is positive. Naturally, a negative test does not rule out the possibility that the individual is a carrier, but it makes it extremely unlikely. In the antenatal setting, screening of mothers to identify carriers followed, when positive, by screening of fathers allows identification of at-risk pregnancies. In the well designed Edinburgh study7 the parents of such pregnancies chose to proceed to fetal genotyping and, when a fetus was diagnosed as having cystic fibrosis, most couples opted for termination of the pregnancy. Widespread adoption of such screening programmes could have a very major effect on the frequency of cystic fibrosis in the future, whilst carrier screening in affected families already provides a high level of reassurance and occasionally assists family planning decisions.
Gene therapy
In theory, cystic fibrosis should be ideal for gene therapy. The main clinical problem is in the airways and the likely target is the surface epithelium. Furthermore, methods of topical delivery to the airway surface are already well developed. Progress was rapid at first: the gene, although large, could easily be inserted into a virus or produced as a plasmid; cellular studies showed that CFTR gene transfer could produce chloride channels which worked and subsequently showed that cystic fibrosis cell lines could be corrected.8 The next steps were the demonstration of relatively efficient gene transfer to the airway epithelium using reporter genes in rodents, followed by partial correction of the disordered airway electrophysiology in CF mice.9 Clinical trials soon followed and, to date, over 150 volunteers with cystic fibrosis have taken part. The results have been both encouraging and frustrating. There is good evidence of low levels of gene transfer and patchy evidence of small changes in ion transport but progress has been hampered by inefficient gene transfer, immunity to viral vectors, and a systemic inflammatory reaction provoked by plasmid DNA.10 ,11 The efficiency of gene transfer is improving as techniques to overcome the biological barriers and cell defence mechanisms are better understood and the gene delivery agents improve. The immune and inflammatory reactions may prove more intractable but eventually will be overcome. However, at this stage no gene therapy methodology has yet approached the desired levels of safety and efficacy needed for large scale clinical trials. Too many enthusiasts promised too much in the early stages and, when they failed to deliver, scepticism set in. Gene therapy is an entirely new technology and it will take another 5–10 years before such a treatment is available. This is neither slower nor faster than the development of any other new drug.
Conclusions
What then has been achieved? Firstly, a mass of new knowledge and understanding; secondly, some tangible benefits in terms of screening and diagnosis; thirdly, some new directions of therapeutic research with reasonable promise of improved treatments to come; fourthly, considerable progress towards gene therapy with likely success within a decade; and finally, the path from gene discovery to clinical application has been cleared and this will speed progress for other genes and other diseases in the future. However, it is worth re-stating that the path is much easier for a single gene defect like cystic fibrosis than it will be for polygenic disorders with major environmental components such as asthma, chronic obstructive pulmonary disease, and lung cancer. When gene linkages are found in these and other diseases, as they certainly will be, do not expect a quick fix.