Article Text

Abstract
BACKGROUND Inflammation, oxidative stress, and recurrent pulmonary infections are major aggravating factors in cystic fibrosis. Nitric oxide (NO), a marker of inflammation, is not increased, however, probably because it is metabolised to peroxynitrite. Exhaled carbon monoxide (CO), a product of heme degradation by heme oxygenase 1 (HO-1) which is induced by inflammatory cytokines and oxidants, was therefore tested as a non-invasive marker of airway inflammation and oxidative stress.
METHODS Exhaled CO and NO concentrations were measured in 29 patients (15 men) with cystic fibrosis of mean (SD) age 25 (1) years, forced expiratory volume in one second (FEV1) 43 (6)%, 14 of whom were receiving steroid treatment.
RESULTS The concentration of exhaled CO was higher in patients with cystic fibrosis (6.7 (0.6) ppm) than in 15 healthy subjects (eight men) aged 31 (3) years (2.4 (0.4) ppm, mean difference 4.3 (95% CI 2.3 to 6.1), p<0.001). Patients not receiving steroid treatment had higher CO levels (8.4 (1.0) ppm) than treated patients (5.1 (0.5) ppm, mean difference 3.3 (95% CI –5.7 to -0.9), p<0.01). Normal subjects had higher NO levels (6.8 (0.4) ppb) than patients with cystic fibrosis (3.2 (0.2) ppb, mean difference 3.8 (95% CI 2.6 to 4.9), p<0.05) and were not influenced by steroid treatment (3.8 (0.4) ppb and 2.7 (0.3) ppb for treated and untreated patients, respectively, mean difference 0.8 (95% CI –0.6 to 2.3), p>0.05). Patients homozygous for the ΔF508 CFTR mutation had higher CO and NO concentrations than heterozygous patients (CO: 7.7 (1.8) ppm and 4.0 (0.6) ppm, respectively, mean difference 3.7 (95% CI –7.1 to –0.3), p<0.05; NO: 4.1 (0.5) ppb and 1.9 (0.7) ppb, respectively, mean difference 2.2 (95% CI –3.7 to –0.6), p<0.05).
CONCLUSIONS High exhaled CO concentrations in patients with cystic fibrosis may reflect induction of HO-1. Measurement of exhaled CO concentrations may be clinically useful in the management and monitoring of oxidation and inflammatory mediated lung injury.
- carbon monoxide
- nitric oxide
- cystic fibrosis
- inflammation
- oxidative stress
Statistics from Altmetric.com
Cystic fibrosis is characterised by recurrent respiratory tract infections leading to damage to the airways, development of bronchiectasis, and progressive airflow obstruction. Infection plays a central role in the progression of the disease and is associated with increased concentrations of several cytokines in the bronchoalveolar lavage fluid, such as interleukin 8 (IL-8), IL-1β, and tumour necrosis factor α (TNF-α),1 ,2 recruitment of neutrophils into the airways, and oxidative stress.3Deficiencies in circulating antioxidants, such as glutathione,4 vitamin E,5 serum albumin,6 and iron,7 have been reported in cystic fibrosis. These deficiencies cause an inability to counterbalance the high production of free radicals, particularly during pulmonary infections, and increased oxidative damage is generated.8
Nitric oxide (NO) may play an important role in regulating airway function and in the pathophysiology of inflammatory airway disease. Exhaled NO has been shown to be a marker of inflammation in asthma9-12 and bronchiectasis.13 However, despite chronic airway inflammation in cystic fibrosis, exhaled NO levels appear to be lower than normal,14-16 possibly because of a reduced expression of the inducible form of NO synthase (iNOS),17 trapping of NO in the mucous layer,18 or because NO is metabolised to peroxynitrite19 ,20 making this measurement of little use for monitoring lung inflammation in cystic fibrosis.
CO is a product of heme degradation by heme oxygenase (HO). Two isoforms of HO have been described—the constitutive HO-2 and the inducible HO-1 which is ubiquitously distributed. The latter is activated by a variety of pro-inflammatory cytokines,21 ,22 oxidants,23NO,24 ,25 endotoxin,26 and reactive oxidant species (ROS),27 and is part of the protective response to oxidative stress.28 ,29
We investigated whether patients with cystic fibrosis have higher exhaled levels of CO than healthy control subjects and whether the levels of exhaled CO and NO are reduced in patients receiving regular inhaled corticosteroids which suppress inflammation in the airways of asthmatic patients.
Methods
PATIENTS
Patients were recruited from the adult Cystic Fibrosis Clinic at the Royal Brompton Hospital. Those colonised withBurkholderia cepacia, methicillin resistantStaphylococcus aureus (MRSA), or with acute chest infection or disease exacerbation were excluded from the study. Twenty nine patients with cystic fibrosis (15 men) of mean (SD) age 25 (1) years, forced expiratory volume in one second (FEV1) 43 (6)% predicted, 15 of whom were receiving treatment with inhaled steroids, and 15 control subjects (eight men) aged 31 (3) years were recruited (table 1). The genotype of 15 patients was investigated by genomic DNA analysis. Fourteen patients were colonised withPseudomonas aeruginosa. None of the patients had clinical signs of pancreatic insufficiency. All patients were life long non-smokers, the smoking status of all the subjects was confirmed by nicCheck (DynaGen Inc, Cambridge, Massachusetts, USA) which detects nicotine and its metabolites in urine. Active and passive smokers (smoke exposure for more than 30 min/day) were excluded from the study.
Mean (SE) characteristics of study subjects
EXHALED CO
Exhaled CO was measured using a modified analyser (EC50-MICRO Smokerlyzer CO Monitor, Bedfont Scientific Ltd, Upchurch, Kent, UK) sensitive to 1–500 parts per million (ppm, by volume) CO adapted for on-line recording. The subjects exhaled slowly from total lung capacity with a constant flow (5–6 l/min) against a resistance of 3 (0.4) mm Hg over 20–30 s.30 Two successive recordings were made and maximal values were used in all calculations. Ambient CO levels were recorded before each measurement.
EXHALED NO
Exhaled NO was measured using a chemiluminescence analyser (model LR2000; Logan Research, Rochester, UK) sensitive to NO from 1 to 5000 ppb (by volume) and with a resolution of 0.3 ppb which was designed for on-line recording of exhaled NO concentration as previously described.31 The analyser was calibrated using certified NO mixtures (90 ppb and 436 ppb) in nitrogen (BOC Special Gases; Guildford, UK). Measurements of exhaled NO were made by slow exhalation (5–6 l/min) from total lung capacity for 20–30 s against a resistance of 3 (0.4) mm Hg.
STATISTICAL ANALYSIS
Comparisons between groups were made by one way analysis of variance (ANOVA) and data were expressed as mean (SE) values with confidence intervals of differences. A p value of <0.05 was considered significant.
Results
EXHALED CO
Exhaled CO levels were increased in patients with cystic fibrosis (6.7 (0.6) ppm) compared with the control group (2.4 (0.4) ppm, mean difference 4.3 (95% CI 2.3 to 6.1), p<0.01; fig 1). Untreated patients had higher CO levels than steroid treated patients (8.4 (1.0) ppm and 5.1 (0.5) ppm, respectively, mean difference 3.3 (95% CI –5.7 to –0.9), p<0.01). There was a tendency for patients colonised with P aeruginosa to have lower levels of exhaled CO than non-colonised patients (6.3 (0.9) ppm and 7.0 (0.9) ppm, respectively, mean difference 0.8 (95% CI –2.3 to 0.6), p>0.05), but this difference was not significant. The homozygous genotype for the ΔF508 CFTR mutation (n = 9) was associated with higher exhaled CO levels than the heterozygous genotype (n = 6) (7.7 (1.8) ppm and 4.0 (0.6) ppm, respectively, mean difference 3.7 (95% CI –7.1 to –0.3), p<0.01, fig 2). There was no difference in disease severity in these two groups of patients.

Concentrations of exhaled carbon monoxide (CO) in normal subjects, patients with cystic fibrosis (CF) on steroid treatment, and untreated patients.

Concentrations of nitric oxide (NO) and carbon monoxide (CO) in the exhaled air of patients homozygous and heterozygous for the ΔF508 mutation.
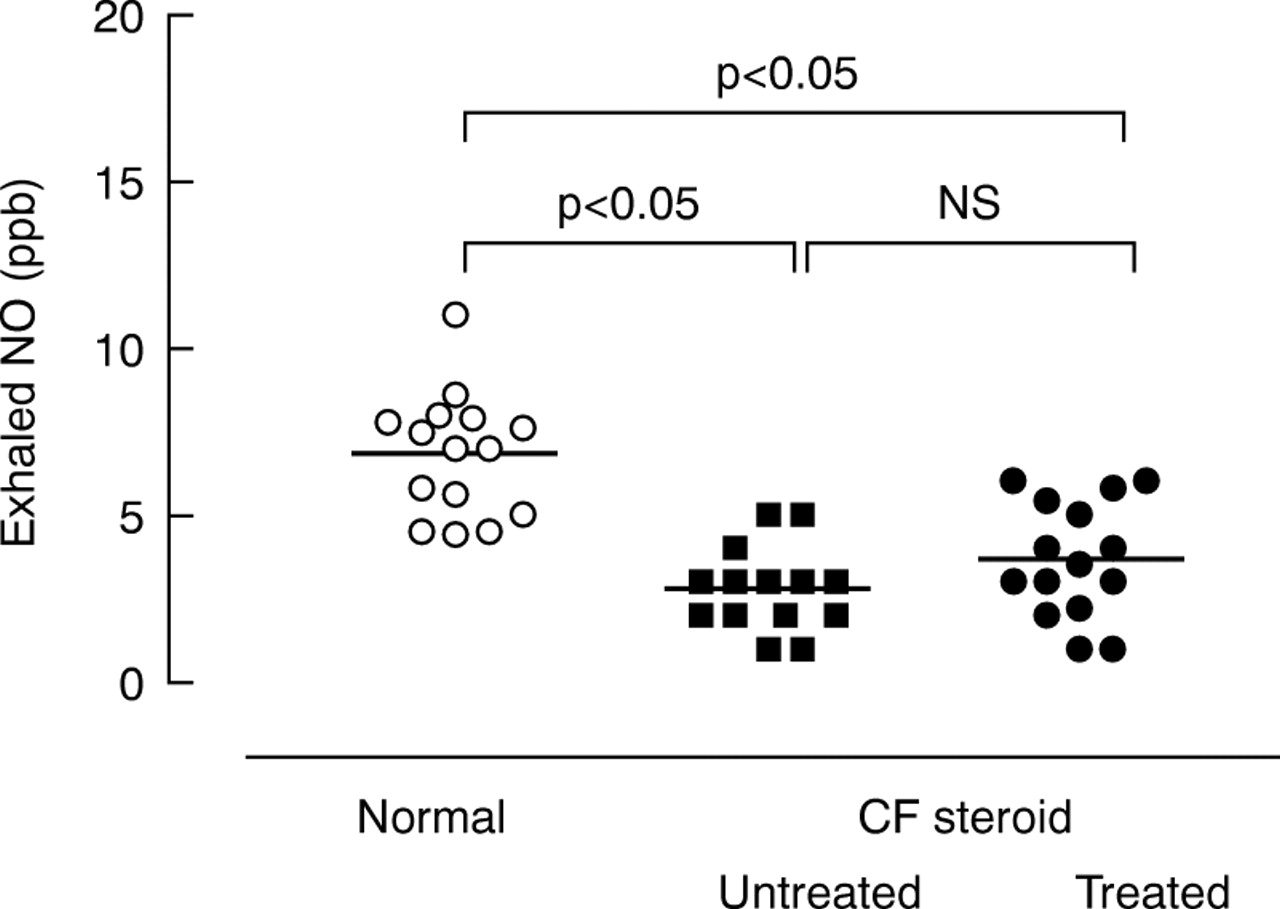
EXHALED NO
Exhaled NO levels were lower in both steroid treated patients (3.6 (0.4) ppb) and untreated patients (2.7 (0.3) ppb) than in the control group (6.8 (0.4) ppb, p<0.05) with mean differences of 3.1 (95% CI 1.6 to 4.6) and 4 (95% CI 2.5 to 5.5), respectively (p<0.05, fig 3). Colonisation with P aeruginosa did not influence exhaled NO levels (2.7 (0.3) ppb and 3.6 (0.4) ppb for colonised and non-colonised patients, respectively, mean difference 0.8 (95% CI –2.3 to 0.6), p>0.05). The homozygous genotype for the ΔF508 CFTR mutation was associated with higher NO levels (4.1 (0.5) ppb) than the heterozygous genotype (1.9 (0.4) ppb, mean difference 2.2 (95% CI –3.7 to 0.6), p<0.05). Neither exhaled NO nor CO concentrations were correlated with FEV1(r = 0.03, p = 0.89 andr = –0.07, p= 0.79, respectively).
{kind=link}
{kind=link}
{kind=link}
Concentrations of exhaled nitric oxide (NO) in normal subjects, patients with cystic fibrosis (CF) on steroid treatment, and untreated patients.
Discussion
We have found that, in patients with cystic fibrosis, exhaled CO concentrations are increased while NO concentrations are decreased compared with normal subjects and that steroid treatment is associated with lower CO concentrations. Furthermore, patients homozygous for the mutation ΔF508 have significantly higher CO and NO levels than heterozygous patients.
Increased levels of reactive oxygen species and oxidative damage have been demonstrated in cystic fibrosis.8 This is possibly due to increased concentrations of oxidation products8 ,32 ,33 and a defect in intracellular oxygen metabolism.34 Glutathione, the major antioxidant in the lung, is reduced both in the epithelial lining fluid of the lung and in the plasma,4 possibly as a consequence of the basic genetic defect.4 The damaging effects of glutathione deficiency are exacerbated by the recurrent infections leading to inflow of activated neutrophils releasing free radicals.35Therefore, circulating and tissue antioxidants are not able to counterbalance the increased generation of free radicals, particularly during bouts of acute pulmonary infection. The increased lung oxidation may stimulate heme catabolism by inducing HO-1 activity as reflected by increased levels of exhaled CO.
Inflammation also has an important role in cystic fibrosis.36 However, despite an increase in cytokines such as IL-1β1 ,2 and TNF-α2 which are known to upregulate the inducible form of NO synthase (iNOS), exhaled NO levels are not increased in patients with cystic fibrosis. The reason for this may be the absence of detectable immunoreactivity for iNOS in lung epithelium.17 Alternatively, high levels of CO may inhibit iNOS activity37 and therefore reduce the levels of exhaled NO. Decreased exhaled NO concentrations may also be due to retention and metabolism of NO within the airways as shown by an increased production of NO metabolites (nitrates and nitrites) in tracheal secretions.19 ,20 Excess NO may be metabolised locally and may activate HO-1 and CO production25 before it has an opportunity to diffuse into the airway lumen.
Heme oxygenase is present in the pulmonary vascular endothelium26 and alveolar macrophages38 and can be upregulated by oxidative stress27 and pro-inflammatory cytokines,21 ,22 increasing the production of CO. The high levels of exhaled CO found in patients with cystic fibrosis may be due to oxidation induced HO-1 hyperactivity and therefore the measurement of exhaled CO may reflect inflammation and oxidative stress. This is further confirmed by the finding of lower CO levels in patients on steroid treatment. In fact, by reducing inflammation,39 attenuating the release of oxidants by inflammatory cells,40 and suppressing pro-inflammatory cytokine production,41 steroids may attenuate HO-1 expression27 and therefore the synthesis of CO.
Cystic fibrosis is characterised by relapsing airway infection and neutrophil recruitment. Considering that neutrophil infiltration of the airways is associated with release of both free radicals8and inflammatory cytokines,1 the use of different markers to separate inflammation from oxidative stress may prove difficult. iNOS is activated by inflammatory cytokines42 whereas HO-1 is induced both by cytokines21 ,22 and oxidants,27 so exhaled NO may reflect inflammation while CO may be a marker of inflammation and oxidative stress. However, NO induces the generation of CO, activating the transcription of the HO-1,25 while high levels of CO reduce NO synthesis, inhibiting iNOS.37 The reciprocal interaction of NO and CO may reduce their sensitivity to detect inflammation and oxidative stress specifically.
Although H2O2 levels are similar in patients with cystic fibrosis and normal subjects,43 other ROS compounds such as superoxide anion may activate HO-1.23Furthermore, H2O2 may still modify HO-1 activity before being metabolised by ROS scavenger such as myeloperoxidase and catalase which are known to be higher in this group of patients.43
The level of exhaled CO is a non-specific marker of oxidative stress in the airways, therefore different levels are not likely to be found in patients colonised with different bacteria. This hypothesis is confirmed by the finding of similar levels of exhaled CO and NO whether or not patients were colonised with P aeruginosa.
Approximately 70% of cystic fibrosis chromosomes carry a phenyalanine deletion at amino acid position 508 (ΔF508).44 We have found higher CO and NO levels in patients homozygous for the ΔF508 mutation. Considering the growing interest in gene therapy in cystic fibrosis, further studies are needed to investigate the role of CO levels in the assessment of effective therapeutic gene delivery or to confirm the diagnosis in patients with borderline sweat tests where more extensive genetic analysis is not available.
Measurement of exhaled CO levels may be a means of detecting and monitoring cytokine mediated inflammation and oxidant stress in the lower respiratory tract and of assessing the efficacy of treatment. Because CO measurement is simple and non-invasive it can be repeated and can be used in children and in patients with severe disease. The increase in exhaled CO levels observed in this study may be of pathophysiological significance. It will be necessary to intervene with specific inhibitors of HO in the future to explore this possibility further.
Acknowledgments
Supported by grants from the University of Milan School of Respiratory Disease and the British Lung Foundation (UK).