Article Text

Statistics from Altmetric.com
Asthma is a complex disease associated with both genetic and environmental factors such as allergens, respiratory tract infections, and atmospheric pollutants. Like rheumatoid arthritis and inflammatory bowel disease, asthma can be regarded as a chronic relapsing inflammatory disorder. Its inflammatory component involves mast cells and eosinophils as the principal effector cells, with dendritic and T cells orchestrating the response.1 ,2 Disease chronicity is associated with tissue destruction and airway wall remodelling which is characterised by epithelial disruption, smooth muscle and microvascular proliferation, and altered matrix deposition.2
Most asthma is associated with atopy, a predisposition to generate immunoglobulin E (IgE) against environmental allergens.3However, only a proportion of atopic individuals develop lower airways symptoms consistent with an asthmatic phenotype, although histological examination of the lower airways of atopic non-asthmatics shows these individuals to have an intermediate degree of eosinophilia and collagen deposition.4 It is therefore tempting to speculate that the development of asthma requires combined inheritance of genes which alter the immune cell response to the environment and, at the same time, render the airways structural and neural regulation susceptible to injury caused by inflammation. Several asthma/atopy associated genes have been identified from linkage and association studies3 ,5 and, as can be seen from table 1, many of the candidate genes are cytokines or cytokine receptors—that is, molecules capable of controlling cell function through regulation of gene expression. Similarly, environmental factors such as ozone are also known to induce gene expression6 ,7 through activation of transcription factors such as NfκB.8 ,9 Thus, it is likely that the primary genetic and environmental causes of asthma have downstream effects on expression of many other genes, and that these ultimately co-operate to determine the asthmatic phenotype (fig1).
Summary of candidate asthma susceptibility genes

Interaction between genes and the environment to produce the asthmatic phenotype.
Consistent with this concept, expression of many genes is known to be altered in the airways of asthmatic subjects. It is noteworthy that this altered profile of gene expression is not confined to inflammatory cells—such as switching of the cytokine profile from Th1 to Th2—but is also evident in airways structural cells. For example, the bronchial epithelium upregulates the expression of a wide range of adhesion molecules, as well as a host of pro-inflammatory cytokines and chemokines relevant to the ongoing inflammatory response of asthma.1 ,2 Subepithelial myofibroblasts are also activated, depositing increased amounts of interstitial collagens types III and V in the lamina reticularis10 which, some studies suggest, correlates with asthma severity as judged by their clinical assessment and lung function.11
The expressed genes in a cell very closely reflect the functional state of that cell and can be used to define cell type, stage of development, and responses to stimuli. Although regulation of protein abundance is not controlled exclusively by regulation of mRNA, virtually all differences in cell type or state can be correlated with changes in the mRNA levels of many genes. Given that measurement of mRNA abundance can be measured readily using a corresponding cDNA sequence, it is now possible to exploit the wealth of information in cDNA databases to ask genome-wide questions through the use of high throughput or large scale experimental methodologies combined with statistical and computational analyses. Thus, the fundamental strategy in functional genomics is to expand the scope of biological investigations from a single gene to studying all expressed genes at once in a systematic fashion.12
So far, studies of gene expression in asthma have been limited to examination of expression at the single gene level using mRNA or protein detection techniques such as RT-PCR, in situ hybridisation or immunohistochemistry. However, a number of powerful methodological approaches have been developed for the study of gene expression at the genome level. Of these, microarray technologies using either cDNAs13 or synthetic oligonucleotides14 ,15deposited onto membranes or glass chips are gaining widespread acceptance as the methods of choice for high throughput expression screening. This technology has already been applied to the study of several diseases including cancer,16 rheumatoid arthritis, and inflammatory bowel disease17; in the two inflammatory diseases the involvement of genes encoding cytokines, chemokines, and matrix metalloproteinases was confirmed, while the novel participation of other cytokines such as IL-3 was also demonstrated. Serial analysis of gene expression (SAGE)18 is another technique that has been developed for use in genome-wide studies; this method is accessible to most molecular laboratories but may be limited by the rather short sequences used for identification purposes. Other methods such as subtractive hybridisation19 and differential display20 have been shown to be useful in identifying novel genes21 and probing expression in complex incompletely characterised genomes; however, they will become less powerful once all human genes have been identified.
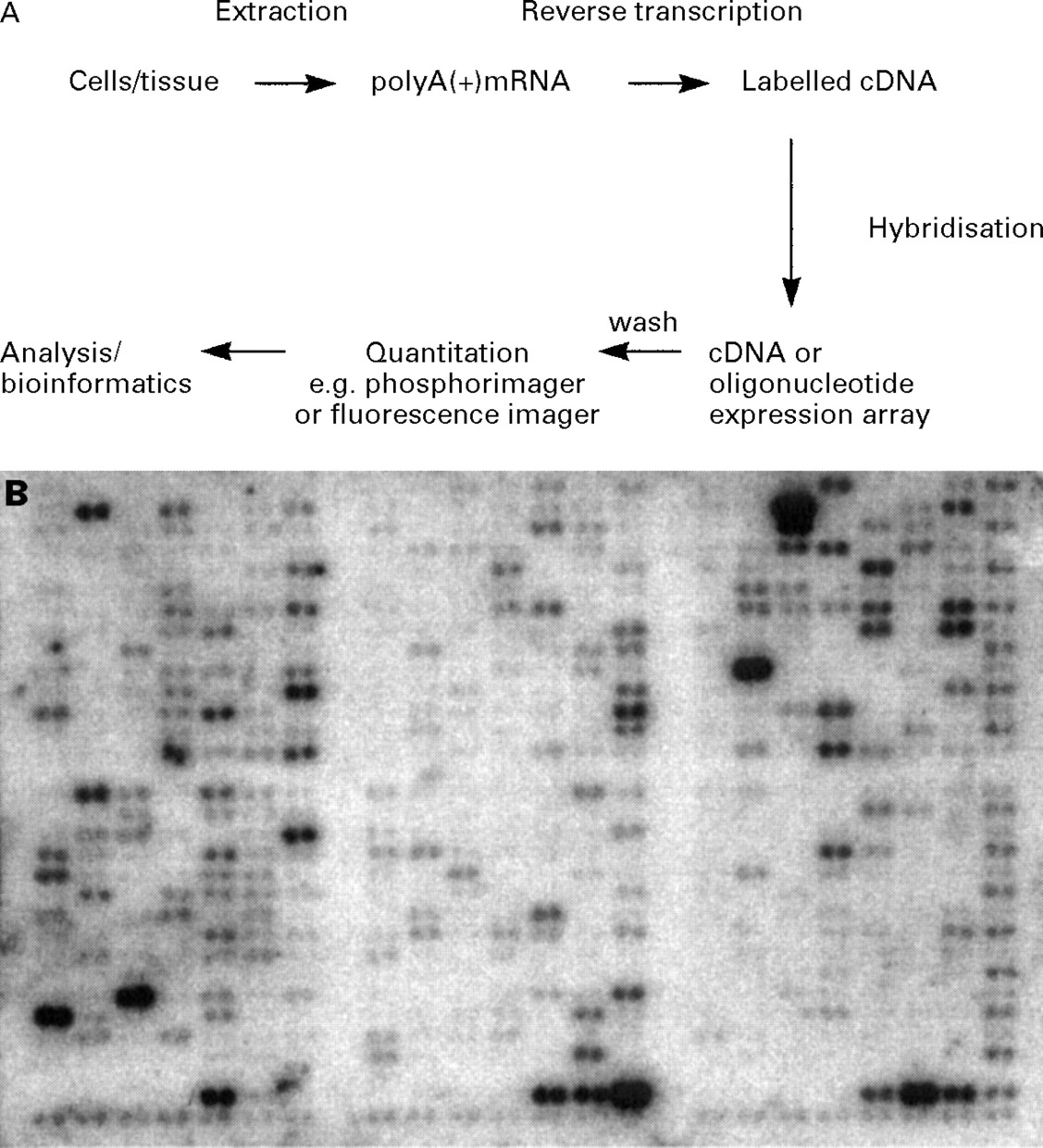
In order to compare patterns of gene expression in two specimens using expression arrays, radioactive or fluorescently labelled cDNA libraries are generated from representative samples of mRNA from each specimen by reverse transcription; these are then allowed to hybridise with the cDNA or oligonucleotide spots of known genes or of expressed sequence tags (ESTs) (usually thousands or tens of thousands of sequences) which have been gridded to form the array (fig 2A). After quantitation and normalisation against a panel of housekeeping genes, relative levels of gene expression between different samples can be computed. In practice, generation of data using expression arrays is relatively straightforward provided that sufficient sample is available for production of high quality mRNA. In order to maximise the information that can be deduced from an array, it is preferable to work with a single cell type. For most in vitro experiments this is not a problem and it is possible to ask well defined questions using well characterised systems—for example, what genes are expressed in a given cell line in response to a particular cytokine? However, when dealing with clinical specimens it may not be possible to obtain a pure cell population; in this case, care must be taken to ensure that the information from the array is not obscured by shifts in the relative numbers of different cell types—for example, differences in the degree of infiltration of inflammatory cells—within the specimen, which will itself alter the types and amounts of genes expressed. This is an essential prerequisite for obtaining data that is both reliable and readily interpretable. While this places some limitations on the types of cells that are available in sufficient quantity for analysis (as a guide, usually around 107 cells are required), in our own experience it is possible to obtain sufficiently pure (>95%) epithelial cells by bronchial brushing using fibreoptic bronchoscopy for use on cDNA expression arrays (fig 2B).
{kind=link}
{kind=link}
(A) Schematic representation of the steps involved in analysis of gene expression using array technology; (B) an AtlasTM array produced using a 32P-labelled complex cDNA probe generated from poly(A+)mRNA extracted from epithelial cells brushed from the airways of a normal non-atopic subject.
Even though most of the sequences contained within an array may be ESTs with no known function, expression arrays can be used to compare expression profiles in normal and disease tissue and so identify candidate genes associated with the disease process. Similarly, analysis of global expression profiles may reveal previously unrecognised subtypes amongst patients and drugs and this may point to mechanisms underlying differences in response to treatment, side effects, etc. Even at this level there is no requirement to know the function of the genes; they may simply serve as markers in cluster analysis. Once implicated, these genes become obvious targets for more detailed study. For in vitro studies, expression arrays offer considerable scope for defining characteristic responses to stimuli, as well as identifying transcriptional targets of a regulatory protein—for example, by engineering its overexpression. Alternatively, mutation of a key regulatory molecule provides a powerful approach to aid dissection and characterisation of regulatory pathways and networks in which it is involved. This latter method can also be used to generate a signature profile for use in a drug discovery programme. Thus, compounds which most closely reproduce the signature profile of gene expression in cells expressing the wild type protein target are most likely to have maximal activity against the target molecule with minimal side effects.
It is to be expected that expression of large numbers of genes will change in the disease situation, particularly in inflammatory diseases such as asthma where a large number of mediators are involved. The challenge therefore is to organise, distribute, extract and interpret data from arrays in an intelligent way. This relies heavily on computational analyses and the availability of appropriate bioinformatics programmes. In the final analysis the key to unravelling such a large body of information will depend on systematically subgrouping the genes on the basis of their known functional activity (if known) or on their global expression patterns in response to given stimuli. Indeed, the information that can be mined from expression arrays will progressively increase, and can be revisited, as more is learnt about the functions of individual genes in specific and more global contexts. As this information accumulates, it will be possible to build a “molecular circuit diagram” of a given cell type which will describe intracellular signalling cascades and the way that they impinge on other pathways to create a three-dimensional interacting network. Although these expression profiles cannot provide unequivocal demonstration of functional interactions between genes, they should enable assignment of subsets of genes indicative of patterns of gene activity or sequential hierarchies. This will facilitate further work aimed at unravelling the complexity of their expression programmes and to trace this back to the underlying cause(s) of diseases such as asthma.
It is conceivable that the rapid improvements in our understanding of genetic factors which determine disease onset and severity may, in the not too distant future, enable treatment to be tailored according to the genetically determined susceptibility of the individual patient. This will, of course, require development of rapid and inexpensive, yet reliable and sensitive, methods for diagnosing the genotype or the phenotype.
In summary, asthma exhibits a variable clinical phenotype which reflects the complex nature of the disease and the contributions from multiple genetic and environmental interactions. Indeed, different genes and different environmental factors may contribute to asthma in different populations. Whatever the underlying causes of the disease, we postulate that they give rise to the asthmatic phenotype by driving expression of distinct sets of interacting genes, some of which are common to all asthmatics, while others may be more restricted in their occurrence. In this review we have considered how functional genomics can help to unravel these complex interrelationships, given the diverse cell types that are involved in the asthmatic phenotype and the involvement of a wide spectrum of cytokines and mediators that can lead to altered profiles of gene expression. This type of approach should provide an insight into the underlying cause of the pathology of asthma and lead to identification of new therapeutic targets.