Article Text

Abstract
BACKGROUND Chronic inflammatory diseases are associated with an increased production of oxidants. Induction of a stress protein, heme oxygenase (HO) HO-1, is a cytoprotective mechanism against oxidative cellular injury. HO-1 catabolises heme to bilirubin, free iron, and carbon monoxide (CO).
METHODS Exhaled CO and sputum bilirubin levels were measured and HO-1 protein expression in airway macrophages was determined by Western blotting in asthmatic patients as levels of oxidants are raised in asthma and may induce HO-1.
RESULTS Exhaled CO was significantly increased in 37 non-steroid treated asthmatic patients compared with 37 healthy subjects (5.8 (95% CI 5.20 to 6.39) ppm vs 2.9 (2.51 to 3.28) ppm; p<0.0001) but was similar to normal in 25 patients who received corticosteroids (3.3 (95% CI 2.92 to 3.67) ppm; p>0.05). In non-treated asthmatic patients more HO-1 protein was expressed in airway macrophages than in normal subjects. Bilirubin levels in induced sputum were also higher than in normal subjects. Inhalation of hemin, a substrate for HO, significantly increased exhaled CO from 3.8 (95% CI 2.80 to 4.87) ppm to 6.7 (95% CI 4.95 to 8.38 CI) ppm (p<0.05) with a concomitant decrease in exhaled nitric oxide levels, suggesting an interaction between the two systems.
CONCLUSIONS Increased exhaled CO levels and HO-1 expression may reflect induction of HO-1 which may be inhibited by steroids. Measurement of exhaled CO, an index of HO activity in non-smoking subjects, may therefore be clinically useful in the detection and management of asthma and possibly other chronic inflammatory lung disorders.
- heme oxygenase-1
- asthma
- exhaled carbon monoxide
- inflammation
Statistics from Altmetric.com
Oxidative stress has been implicated in the pathogenesis of many diseases including atherosclerosis, carcinogenesis, and chronic inflammatory disorders such as asthma, chronic obstructive pulmonary disease (COPD), rheumatoid arthritis, and psoriasis.1-5In these chronic diseases reactive oxygen species (ROS)—including the superoxide anion, hydroxyl radicals, and hydrogen peroxide (H2O2)—are produced by activated immune and inflammatory cells. These mediators amplify and perpetuate the disease state by the oxidation of nucleic acids, proteins, and membrane lipids.6 Organisms have evolved both non-enzymatic and enzymatic antioxidant defence systems to detoxify these harmful oxidants. One of these defence mechanisms is the induction of a stress response protein, HO-1.6 ,7 HO-1 catalyses the initial and rate limiting step in the oxidative degradation of heme to bilirubin. The enzyme, utilising NADPH and molecular oxygen, cleaves a meso carbon of the heme molecule producing biliverdin, free iron, and CO.8 Biliverdin is then converted to bilirubin by bilirubin reductase. Bilirubin itself is an antioxidant and CO has several biological activities including stimulation of guanylate cyclase.9 HO-1 induction therefore has been suggested as a protection against oxidant mediated cellular injury.10Several mediators which are also released during chronic inflammation including cytokines, ROS, and nitric oxide (NO) are able to induce HO-1 expression under experimental conditions.6 ,7 Despite these lines of evidence implicating a potential induction and role for HO-1 in human diseases associated with oxidative stress, its expression and activity have not been determined in these conditions.
Asthma is a chronic inflammatory disease of the airways with increased ROS and nitric oxide (NO) production and raised levels of several inflammatory mediators in the airways, all of which may be potential inducers of HO-1 expression.3 ,11
Increased levels of exhaled CO have recently been demonstrated in patients with asthma not treated with corticosteroids, and changes in CO concentration were significantly related to those in the eosinophil cell counts in sputum.12 We postulated therefore that HO-1 enzyme would be expressed more intensively and levels of its end products, CO and bilirubin, would be higher in the airways of asthmatic patients than in normal subjects. Glucocorticosteroids, which control inflammation in asthmatic airways, may inhibit the expression of HO-1 and therefore may result in a decreased concentration of exhaled CO. We have determined the levels of HO-1 protein expression in macrophages obtained from induced sputum, measured levels of bilirubin in sputum supernatant, and CO in exhaled air in normal and asthmatic subjects. As active and passive smoking influences levels of exhaled CO and may interfere with its endogenous production, we therefore recruited only non-smoking individuals and tested non-smoking status by determining the levels of nicotine and its metabolites in urine.
Methods
SUBJECTS
Non-smoking subjects were recruited from volunteers taking part in other studies and from outpatients at the Royal Brompton Hospital. None of the controls (20 men and 17 women of mean age 33 (95% CI 27.5 to 38.6) years) had a history of respiratory or cardiovascular disease or was receiving long term medication. Asthmatic patients were atopic and had documented reversible airway obstruction and airway hyperresponsiveness (methacholine PC20 <8 mg/ml). They were stable for at least two weeks before study. One group received either no regular treatment or inhaled β2 agonists alone (20 men and 17 women of mean age 32 (95% CI 26.7 to 37.3) years) and the others received regular inhaled steroids (beclomethasone dipropionate or budenoside 400–1600 μg daily; 11 men and 14 women of mean age 36 (95% CI 30.3 to 41.5) years). Spirometric tests showed a mean forced expiratory volume in one second (FEV1) of 94 (95% CI 87.9 to 100.1)% predicted and 77 (95% CI 70.0 to 83.9 CI)% predicted for the non-steroid treated and the steroid-treated groups, respectively. Subjects were tested by NicCheck I (DynaGen Inc, Cambridge, Massachusetts, USA), which determines the levels of nicotine and its metabolites, to ensure non-smoking status and active and passive smokers (smoke exposure for more than 0.5 hour/day) were excluded from the study. The study was approved by the ethics committee of the Royal Brompton Hospital and informed consent was obtained from all subjects.
EXHALED CO
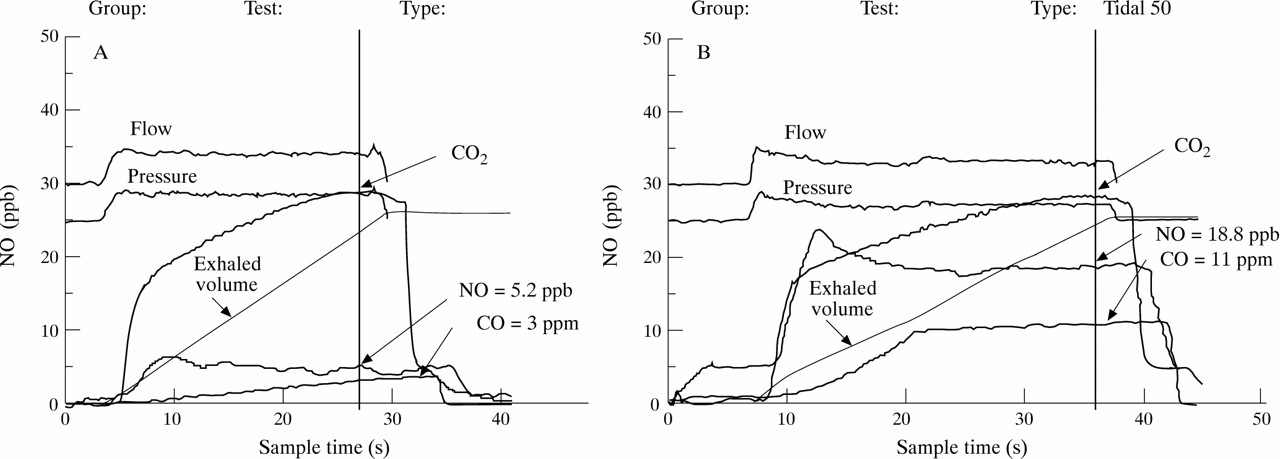
Exhaled CO was measured by a modified analyser (EC50-MICRO Smokerlyzer CO monitor, Bedfont Scientific Ltd, UK) sensitive to CO from 0 to 500 parts per million (ppm, by volume), adapted for online recording of CO concentration and integrated with a chemiluminescence analyser (LR 2000, Logan Research, Rochester, UK) to control exhalation parameters. The subjects exhaled slowly from functional vital capacity with a constant flow (5–6 l/min) against resistance (3 (0.4) mm Hg) over 20–30 seconds into the analyser. Two successive recordings were made and mean values were used in all calculations. Ambient CO levels were recorded before each breath. Exhaled NO was measured as described previously.13
To show that baseline levels of CO can be increased by influencing heme oxygenase (HO) activity, six subjects inhaled solution of an active substrate analogue, hemin (Sigma Chemical Co, Poole, UK) (2 ml of 10–4 M). Levels of exhaled CO and NO were measured for four hours since changes in CO levels may interact with NO production.
INDUCED SPUTUM
HO-1 protein expression was measured in airway macrophages obtained from induced sputum samples. Sputum was induced from nine non-steroid treated asthmatic and seven normal subjects. Subjects inhaled 3.5% saline solution at room temperature nebulised by an ultrasonic nebuliser (DeVilbiss 99, De Vilbiss, Heston, UK) for 15 minutes. Subjects were encouraged to cough deeply after five minutes and at five minute intervals thereafter. Sputum was coughed into polypropylene containers and were kept at 4°C for not more than two hours before further processing. The volume of the samples was recorded. The sample was diluted with phosphate buffered saline containing 10 mmol/l dithiotreitol and gently vortexed at room temperature. The samples were then centrifuged at 400g for 10 minutes and the cell pellet thus formed was resuspended. The supernatant was stored at –70°C for subsequent assay for bilirubin concentration. The cell pellet was washed twice with Hank’s balanced salt solution (HBSS) without Ca2+ and Mg2+. The cells were stained with Kimura stain and macrophages counted using a haemocytometer. The macrophages were resuspended in RPMI-1640 media containing 10% (v/v) fetal calf serum, 2 mM glutamine 100 μg/ml penicillin, and 100 u/ml streptomycin. Macrophages were seeded at a density of 1 × 106 cells/well on a six well plate and allowed to adhere for 90 minutes in a humidified incubator at 37°C and 95% (v/v) air, 5% (v/v) CO2, after which the non-adherent cells were removed by washing with HBSS. The adherent cells were scraped off the plate and identified as macrophages by staining with Kimura stain. This process resulted in the recovery of approximately 50% of the plated cells and there were no differences in the recovery between each group of subjects. The resulting cell pellet was stored at –20°C until required.
BILIRUBIN ASSAY
The total bilirubin concentration in induced sputum was measured automatically by a time endpoint diazo method (Sinchron CX Systems, CX7 Delta Analyzer, Beckman Institute, UK) in the Department of Biochemistry, Royal Brompton Hospital.
HO-1 PROTEIN EXPRESSION
The gel system we use will not allow for all the samples to be run on the same gel, therefore we have used a blot that is representative of three separate gels. The HO-1 antibody used here is available commercially and has no cross reactivity with HO-2. The cell pellets were homogenised in 50 mM Tris/HCl pH 7.4 containing 0.25 mM EDTA, 0.5 mM PMSF, 5 μg/ml antipain, 5 μg/ml leupeptin, and 5 μg/ml benzamidine. Protein was determined using the BIORAD protein assay. The proteins were solubilised by boiling in SDS-PAGE sample buffer (0.0625 mM Tris/HCl pH 6.8 containing 10% v/v glycerol, 1% w/v SDS, 1% w/v β-mercaptoethanol, and 0.01% w/v bromophenol blue). The proteins (10 μg per lane) were resolved by electrophoresis in 12% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Equal protein loading was determined by staining the blot with 0.1% (w/v) Porcean S in 5% (v/v) acetic acid. The nitrocellulose was blocked overnight at 4°C in 10% w/v dried milk protein in PBS containing 0.05% v/v Tween-20. The blots were washed in PBS containing 0.05% v/v Tween-20 and incubated for one hour in the presence of rabbit-anti-heme oxygenase-1 antibody (1:7500) (Affiniti Prod. Ltd, Exeter, UK). The blots were washed extensively and then incubated for one hour with anti-rabbit IgG conjugated to horse radish peroxidase (1:4000). The blots were washed extensively again and the bands were visualised using ECL (Amersham, Amersham Place, UK).
ANALYSIS OF DATA
The 95% confidence intervals (CI) were calculated with standard algorithms. Comparisons between groups were made by the Student’s t test. Significance was defined as p<0.05.
Results
EXHALED CO
Exhaled CO was detectable in all subjects (fig 1). Measurements in normal subjects (n = 33) were reproducible, the variation between readings on separate days being 5.3%. Ambient air concentrations of CO (0–2 ppm) did not affect exhaled CO readings as there was no difference in CO levels after inhalation of certified CO free air (BOC, UK) compared with room air in four normal and five asthmatic subjects. The exhaled CO concentration was 2.9 (95% CI 2.51 to 3.28) ppm in normal non-smoking subjects.

Representative original traces of expired CO and NO in (A) a normal subject and (B) an asthmatic patient.
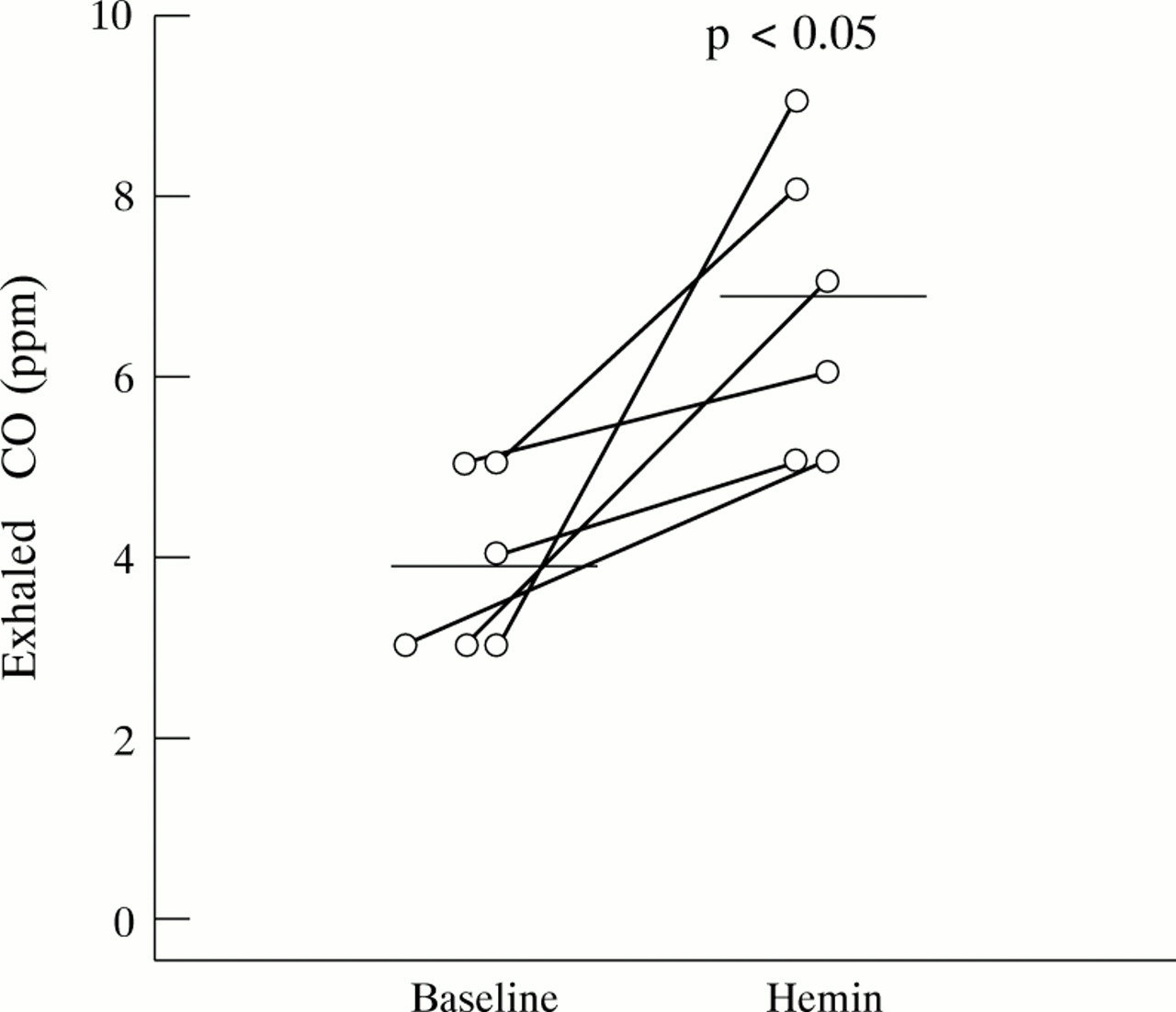
Hemin inhalation significantly increased expiratory CO concentrations from 3.8 (95% CI 2.8 to 4.87) ppm to 6.7 (95% CI 4.95 to 8.38) ppm (p<0.05) with a concomitant decrease in exhaled NO concentrations from 5.4 (95% CI 4.22 to 6.57) ppb to 3.2 (95% CI 1.84 to 4.60) ppb (p<0.01) in six normal subjects (fig 2).
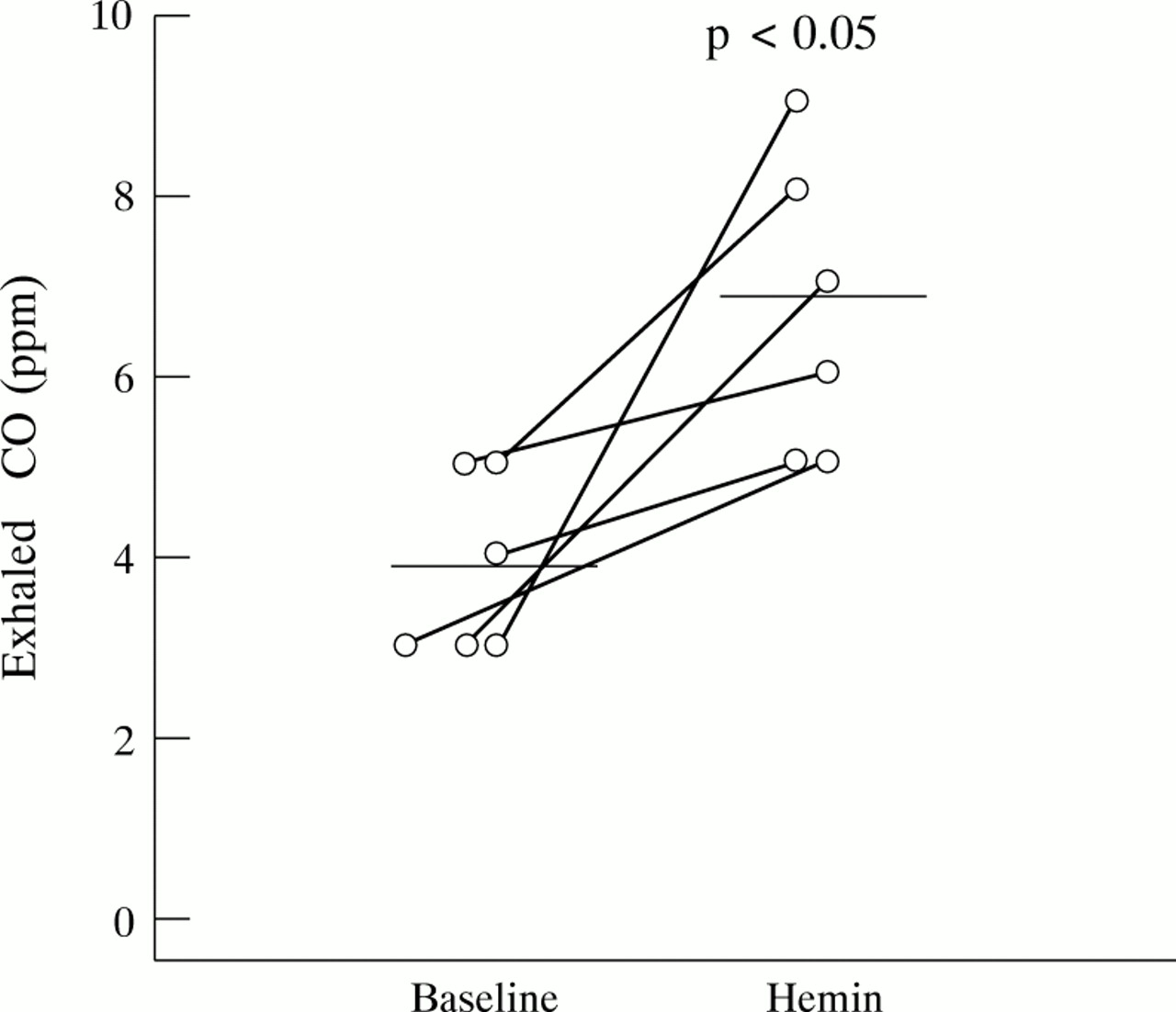
Effect of hemin on exhaled CO levels in normal subjects (n = 6).
In asthmatic patients not receiving corticosteroids the CO levels were significantly higher than normal (5.8 (95% CI 5.20 to 6.39) ppm; p<0.0001) whereas in asthmatic patients receiving inhaled corticosteroids exhaled CO was significantly reduced compared with values in untreated subjects and did not differ significantly from those of normal subjects 3.3 (95% CI 2.92 to 3.67) ppm (fig 3).

Exhaled CO concentrations in normal subjects (n = 37), asthmatic patients not on inhaled steroids (n = 37), and asthmatic patients on inhaled steroids (n = 25).
SPUTUM BILIRUBIN LEVELS
Bilirubin was detectable in each of the sputum samples and its concentration was increased in nine patients with asthma compared with seven normal subjects (5.8 (95% CI 4.61 to 6.89) μM vs 3.6 (95% CI 2.82 to 4.40) μM; p<0.05).
HO-1 EXPRESSION
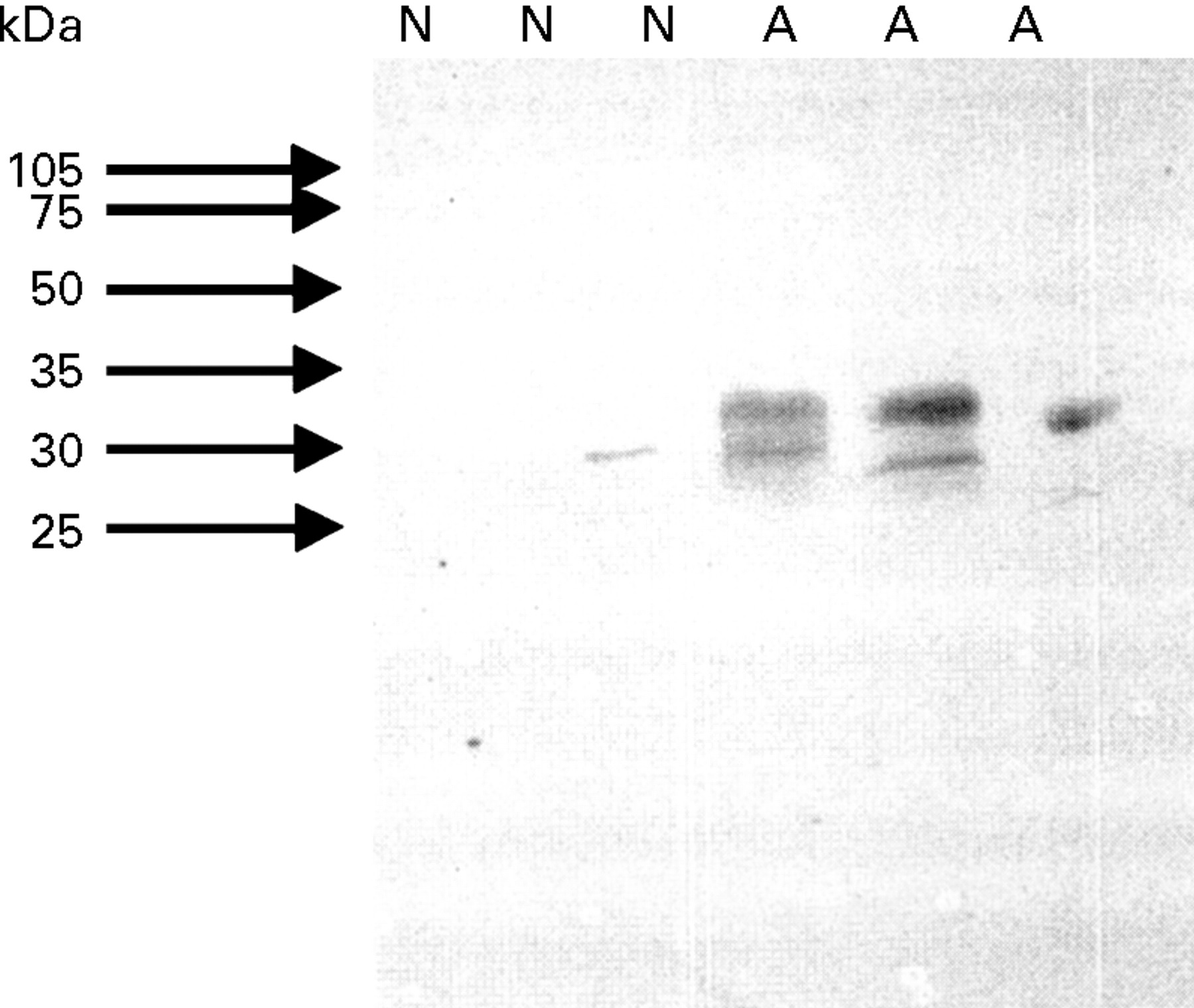
Western blot analysis showed that HO-1 is expressed at a higher level in airway macrophages obtained from induced sputum of asthmatic patients (n = 6) compared with normal subjects (n = 6) (fig4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Western blot analysis of HO-1 in airway macrophage lysate (10 μg protein/lane); N = samples from control healthy subjects; A = samples from asthmatic patients.
Discussion
This study shows that raised levels of CO in exhaled air of asthmatic subjects are associated with increased expression of HO-1 in induced sputum macrophages and increased levels of bilirubin in sputum compared with healthy subjects, and that exhaled CO may be modified by corticosteroid treatment. Inhalation of hemin significantly increased the levels of exhaled CO in normal and asthmatic subjects. Detection of exhaled CO as an index of heme oxygenase activity may therefore reflect oxidative stress and could be a simple non-invasive tool for monitoring airway inflammation.
These results confirm the early observation12 that exhaled CO levels are raised in asthma, reduced by inhaled corticosteroids, and that ambient air CO concentrations (at levels of 0–2 ppm) do not affect exhaled CO.
Exhaled CO and sputum bilirubin concentrations were significantly higher than normal in asthmatic patients who were not receiving inhaled steroids. In asthmatic airways induction of HO-1 may lead to the high exhaled CO concentrations observed in the present study. Increased HO-1 protein expression may be due to the induction of the enzyme by inflammatory mediators present in the asthmatic airways. Many cytokines and oxidants involved in asthmatic inflammation—including interleukins, tumour necrosis factor α, interferon-γ, NO, and H2O2—are able to induce HO-1 expression in cell lines and tissues under experimental conditions.6 ,7
This study, however, is the first to show an increase in HO-1 expression in vivo in patients. The normal exhaled CO levels in steroid treated patients suggest that inhaled steroids may inhibit HO-1 expression in the airways.
The cellular source of CO in asthmatic patients is not known although our present study would implicate airway macrophages. These cells are known to be activated in asthma and to produce superoxide anions after activation, which may explain the increased HO-1 expression in these cells.3 HO-1 may be induced in various other superficial cells in the respiratory tract including epithelial cells and infiltrating inflammatory cells.14 The induction of HO-1 has been implicated as an antioxidant defence mechanism. By degrading heme, a pro-oxidant contained in a multiplicity of intracellular proteins, and by generating bilirubin, an antioxidant which can scavenge hydroxyl radicals in vitro as efficiently as α-tocopherol, induction of HO-1 may provide antioxidant protection.9 ,15CO, a by-product of HO activity, activates guanylate cyclase and it is a putative neurotransmitter.16 However, when produced in high local concentrations by inducible HO, CO may have pro-inflammatory effects since it is also a potent vasodilator and may increase plasma exudation from airway vessels.17 Free iron, another end product of HO-1, can act as a catalyst in the formation of ROS and through this mechanism it may have inflammatory effects; however, it also induces ferritin expression which may serve to restrict iron from participation in the Fenton reaction, thereby reducing the oxidant burden of the cell.10 ,18
The induction of HO-1 has been shown to participate in resolution of acute inflammation under experimental conditions and it has been suggested that it may play a cytoprotective role in heme and oxidant induced cellular injuries.19 These observations raise the possibility that overexpression of HO-1 may be beneficial in the treatment of chronic inflammatory diseases. Measurement of exhaled CO, an index of HO activity, may be a simple method of detecting and monitoring cytokine mediated inflammation in the respiratory tract and of assessing anti-inflammatory treatments. As CO measurement is simple and non-invasive, it is repeatable and is possibly useful in children and patients with severe disease who may not tolerate more invasive investigation. Further studies are needed, however, to relate expired CO and sputum bilirubin levels to the extent of inflammation and to explore this approach in other inflammatory lung diseases. We speculate that there may be a sequential induction of enzymes that include cyclooxygenase-2 and inducible nitric oxide synthase, followed by increased expression of enzymes with anti-inflammatory effects, including HO-1. The close inverse relationship between the regulation of CO and NO production is reflected by the reduced exhaled NO concentrations after increasing exhaled CO levels.
In conclusion, the findings of this study implicate a direct involvement of HO-1 in asthmatic inflammation. The presence of HO-1 in the airway macrophages, a cell type closely associated with chronic inflammation, would support the concept of this enzyme as a modulator of the chronic inflammatory response. Measurement of exhaled CO may be clinically useful for monitoring oxidant mediated pulmonary diseases.
Acknowledgments
IH was supported by the European Respiratory Society and the Hungarian National Scientific Research Foundation (OTKA-F017050), LED by the National Asthma Campaign (UK), SAK by the British Lung Foundation, and PP by the University of Milan.
References
Characteristics of subjects