Article Text

Statistics from Altmetric.com
Prior to the discovery of the tubercle bacillus, the observation that tuberculosis frequently occurred in many members of the same family convinced physicians that it was a hereditary disease.1 However, in 1882 when Robert Koch demonstrated that tuberculosis was caused by a microorganism,2attention turned to the importance of the pathogen and the genetic constitution of the host was largely ignored. It is estimated that only 10% of those who become infected withMycobacterium tuberculosis will ever develop clinical disease,3 and in only a few cases is there an obvious identifiable risk factor such as diabetes, advanced age, alcohol abuse, HIV infection, or corticosteroid usage. Why then do some people succumb to the disease when most of the population can successfully fight off the tubercle bacillus? Clearly, althoughM tuberculosis is necessary, it is not sufficient. Determining why only some individuals are susceptible to tuberculosis is important as this will give us insight into which components of disease pathogenesis are important and hopefully assist the development of new treatment or prevention strategies.
It is a common misapprehension that death from multifactorial diseases such as cancer and cardiovascular problems are influenced by genetic factors, but that death from infection is due to a bad environment or simply bad luck. In fact, a study of 960 adoptees in Denmark concluded that the genetic component of susceptibility to premature death from infectious diseases is greater than for cancer or cardiovascular causes.4 In 1949 the pioneering geneticist J B S Haldane recognised that infectious diseases have been the main agent of natural selection during the past 5000 years.5 Organisms which are widespread in the human population and which exert a high mortality will exert the most pronounced evolutionary effects.6 In the early part of the 19th century it is estimated that, in Western Europe, tuberculosis was responsible for between one quarter and one fifth of all deaths,7 and during the Industrial Revolution this figure may have been even higher.1 We should therefore expect that M tuberculosis will have exerted as large an effect upon maintenance of genetic polymorphism throughout the human genome as hasPlasmodium falciparum. However, although many gene variants which confer innate resistance to malaria have been identified,8 this has proved much more difficult for tuberculosis. This difference is probably due to the geographical variation in the prevalence of malaria which has resulted in malaria resistance mutations clustering in certain ethnic groups, thus facilitating their identification. As tuberculosis is a global problem it has not been possible to identify tuberculosis susceptibility or resistance genes using interethnic comparisons. Although it has proved difficult to identify tuberculosis susceptibility genes, there is convincing evidence that host genetic factors are important in determining the outcome of infection.
Evidence linking tuberculosis with host genetics
Over 100 years ago it was recognised that there are racial differences in susceptibility to tuberculosis.9-11 This was most strikingly observed in the Qu’Appelle Indian Reservation, Saskatchewan in 1890. When the population first became exposed to tuberculosis the annual death rate from tuberculosis reached almost 10% of the population. After 40 years over half the Indian families were eliminated and the annual tuberculosis death rate had fallen to only 0.2%, presumably related to the strong selection pressure against tuberculosis susceptibility genes.6 It has been suggested that a population’s resistance to tuberculosis is determined by its previous history of exposure.12 ,13 In general, black populations have greater susceptibility to tuberculosis than Caucasians, perhaps because tuberculosis has been endemic in Europe for a much longer period and has therefore been a more significant evolutionary force.1 Black populations have higher rates of tuberculosis and are also more likely to develop the more fulminant forms of the disease.1 ,14 Although many physicians would attribute these racial differences to environmental factors, there is evidence that the difference is largely due to genetic susceptibility. Stead and coworkers found that, among over 25 000 tuberculin negative nursing home residents, black subjects were twice as likely to become infected with tuberculosis as white subjects living in the same environment, and that this could not be explained by any identifiable environmental factors.15
Probably the most convincing evidence that genetic factors are important in tuberculosis susceptibility comes from studies of twins. These studies compare how often the monozygous (MZ) or dizygous (DZ) twin of an index case of tuberculosis is also affected by the disease. A higher concordance for disease among MZ than among DZ twins suggests that genetic factors are important in disease susceptibility. Twin studies in tuberculosis have consistently found much higher disease concordance among MZ than DZ twins, which suggests that, even within ethnic groups, host genetic factors exert a major influence on tuberculosis susceptibility.6 ,16-18
Attempts to identify the actual genes involved in host susceptibility to tuberculosis have until recently focused on the human leucocyte antigen (HLA) system. Associations have been found between the class I HLA antigens A10 and B819 and with the class II antigen DR2.19-21 However, these associations have not been consistently demonstrated22 and could account for only a small part of the significant genetic component in tuberculosis susceptibility identified by the twin studies.
Animal models of tuberculosis susceptibility
Animal models of tuberculosis have been widely used to attempt to further our understanding of the pathogenesis of the disease. In 1882 Koch used the guinea pig to demonstrate that M tuberculosis was the causative agent of human tuberculosis.2 This animal is extremely sensitive to tuberculosis and develops disease which is clinically and histologically similar to that found in humans.23 However, there has been little research on the genetic susceptibility of inbred guinea pig strains to tuberculosis. Evidence that different inbred animals exhibit strain-specific susceptibility to tuberculosis has mostly accumulated from work on rabbits and mice.
The classic studies of Lurie found that inbred rabbit families exhibited two patterns of disease following infection with virulentM bovis. Resistant rabbits developed cavitary pulmonary disease resembling that found in immunocompetent adult humans and susceptible rabbits developed disseminated haematogenously spread disease resembling that found in infants and immunocompromised subjects.24 ,25 When infected with human type M tuberculosis the resistant rabbit families were found to inactivate more tubercle bacilli than the susceptible rabbits.26 The inheritance pattern of the practically all-or-none responses observed led Lurie to conclude that resistance was largely genetically determined.26
Studies on inbred strains of mice have identified two distinct phenotypes designated Bcg s andBcg r which are, respectively, sensitive and resistant to infection with intracellular pathogens such as Mycobacterium,Salmonella andLeishmania.27 Positional cloning has identified the gene responsible for theBcg phenotype to be the natural resistance associated macrophage protein (Nramp) gene on mouse chromosome 1.28 A single glycine to aspartic acid base substitution at codon 169 within the second predicted transmembrane domain of Nramp has been shown to be responsible for susceptibility to all three pathogens.28 An Nramp gene disrupted “knock-out” mouse was produced and shown to have the sameBcgs phenotype as theNramp aspartic acid169homozygous mouse.29 A transgenic mouse in which the wild-type glycine169 allele was transferred onto the background of a mouse homozygous for aspartic acid at position 169 was shown to have the Bcgr phenotype.30 These experiments confirmed thatNramp, and not another closely linked gene, is responsible for the Bcg phenotype. Mice with targeted gene disruptions of the genes encoding interferon-γ, the interferon-γ receptor and β2-microglobulin have also been found to be extremely sensitive to Mycobacterium.31-33 These mice are analogous to humans with single gene disorders such as adenosine deaminase deficiency, chronic granulomatous disease, and interferon-γ receptor deficiency who are susceptible to disseminated BCG and atypical mycobacterial infections.34-36 As yet, none of these genes has been found to contain a common functional variant which might explain host variability in susceptibility to tuberculosis in humans. They are a useful starting point for any tuberculosis susceptibility gene hunt and the Nramp gene is a particularly strong candidate.
Investigating human tuberculosis susceptibility by linkage and association
Family based genetic linkage studies and population based case-control (or association) studies are two complementary strategies used to identify the genes involved in a multifactorial disease. Each approach has its advantages and limitations. Linkage analysis can be used to screen the entire human genome for regions of chromosomes segregating with disease.37-46 This method is comprehensive and should locate any gene which exerts a major effect on disease susceptibility, but it has relatively low power and will fail to detect genes exerting only a moderate effect on risk of disease.37 ,47 For example, if a disease susceptibility allele exerts a twofold risk of disease compared with the wild-type allele, between 2500 and 300 000 families (depending on allele frequencies) would need to be typed to detect linkage to this gene, numbers which are not practically achievable.47Association studies have much greater power but, as association is detectable over much smaller regions than linkage, many more markers would need to be typed to conduct a genome wide association study. This is not possible with current technology so at present association studies are limited to the investigation of candidate genes and regions identified in linkage studies. Linkage studies can therefore ensure that genes exerting large effects on disease susceptibility have not been missed but will fail to identify many genes exerting moderate effects on disease risk. Association studies can detect genes which exert smaller effects but, as they are not comprehensive, the possibility that the most important genes have been overlooked cannot be excluded. Any serious attempt to identify disease susceptibility genes should utilise both strategies.
Genome wide linkage studies
Linkage analysis of complex traits is most widely carried out using affected sib pairs, a method originally described by Penrose.48 Families are collected which include two full siblings who share the disease phenotype and their parents, who preferably do not have the disease. Three families of this type are shown in fig 1. If the family is typed for a fully informative polymorphic DNA marker then the number of parental alleles which the offspring share can be determined. When the marker locus is not located in the same chromosomal region as a disease susceptibility gene, the marker alleles are inherited independently of the disease phenotype. This results in offspring sharing two parental alleles identical by descent (ibd) in 25% of families, one allele ibd in 50% of families, and zero alleles ibd in 25% of families. If the marker locus is located close to a disease susceptibility gene—they are linked—then there should be a significant excess of families where two alleles are shared ibd and fewer families where offspring share zero alleles compared with the expected frequencies. This non-parametric system of analysis makes no assumptions about the mode of inheritance of the disease, which is an advantage for complex diseases where this information is usually not available.

The numbers below each family member represent alleles at a marker locus, which is not linked to the disease. In the first family both offspring inherit allele 1 from the father and allele 3 from the mother and thus share two alleles identical by descent (ibd). In the second family they have inherited the same paternal allele but different maternal alleles. In the third family they have inherited different alleles from both parents. If a large number of families are typed the expected frequencies of the three inheritance patterns are 25%, 50%, and 25%.
Generally at least 100 families are required to carry out a genome screen on a multifactorial disease, using around 300 highly informative microsatellite markers spaced at approximately 10 centiMorgan intervals (1 cM represents a 1% probability of recombination during meiosis). Microsatellites are dinucleotide repeats of a (CA)nsequence which are highly polymorphic due to variation in the number of copies of the repeat at a single locus throughout the population.49 The identification of thousands of microsatellites50 ,51 and the development of semi-automated fluorescence based typing systems52-54have revolutionised linkage studies. Polymerase chain reaction (PCR) primers are labelled with one of three fluorescent dyes and microsatellite marker loci PCR products are separated by size using polyacrylamide gel electrophoresis. Using a 373 or 377 sequencer (Perkin-Elmer), a laser detects the three dyes and a fourth dye used to label a size standard. Computer software (Genescan 672 and Genotyper) is used to compare the time taken for the fluorescently labelled PCR product to reach the laser with time taken by the size standard to accurately calculate the size of microsatellite PCR products from a single locus.52 ,53 Using sets of microsatellites which do not overlap in size, it is possible to run PCR products with a single dye label from up to eight microsatellite loci in a single gel lane. Using three dyes up to 24 microsatellite loci can be analysed in a single lane or 864 genotypes per gel.53 Figure 2 shows the large number of microsatellites which can be typed in a single gel lane and fig 3 demonstrates how Genotyper software is used to focus on a single microsatellite and trace the inheritance of alleles within a family.

Nineteen microsatellite loci typed using a single lane on an ABI 373 sequencer. Each row shows the microsatellites labelled with the FAMTM (blue), TETTM (green) and HEXTM (yellow) fluorescent dyes. The bottom lane is the TAMRATM (red) size standard.
{kind=link}
{kind=link}
{kind=link}
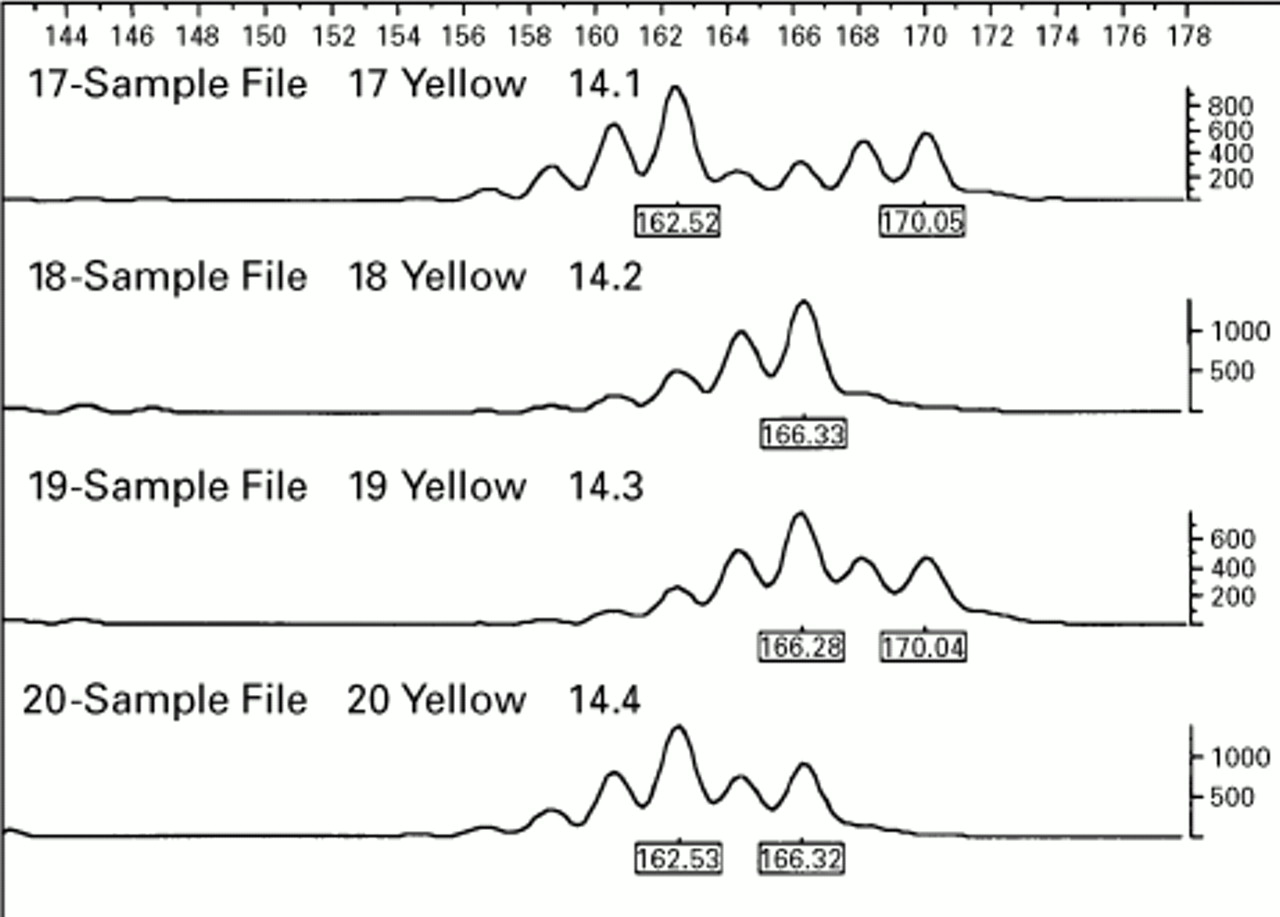
Inheritance of alleles from a single microsatellite locus in a sib pair family: individual: 14.1 mother, 14.2 father, 14.3 sib 1, 14.4 sib 2. The offspring have inherited different maternal alleles but as the father is a homozygote it cannot be determined whether or not they have inherited the same paternal allele. Note that each allele is followed by shorter “stutter” bands produced by PCR slippage.
Using this technology we have recently completed a whole genome screen with 282 microsatellite markers on 92 sibling pairs from the Gambia and South Africa and have identified four regions which show evidence of segregation with tuberculosis (Bellamy et al, unpublished data). However, due to the large number of markers typed false positive, linkages to regions which do not include a disease susceptibility gene can arise. To overcome this problem many genome screens have employed two stages.37 ,38 ,42 ,43 ,45 ,55 In the first screen all markers are typed in order to cover the whole genome. Markers which show evidence of linkage to disease are then typed in a second set of families. This approach minimises the risk of false positive linkages occurring. Currently more tuberculosis sib pair families from Africa are being studied in order to confirm the linkages detected.
Association based candidate gene studies
Association studies involve typing a genetic polymorphism in a large number of unrelated individuals with the disease of interest and a group of healthy ethnically matched controls. Marked differences in genotype frequency between the two groups suggest either that (a) the polymorphism predisposes to the disease, (b) the polymorphism is in linkage disequilibrium with a disease susceptibility gene, or (c) there is a confounding factor such as poor ethnic matching between the cases and controls. In order to have adequate power to detect moderate genotype relative risks it is necessary to type several hundred individuals47 and many case-control studies examine grossly inadequate numbers. In a current tuberculosis case-control study in Gambians we routinely type over 800 unrelated individuals. Linkage disequilibrium is usually only present over distances of less than 1 cM, a much shorter distance than family based linkage analysis can be used for. Association studies are therefore useful in attempting to localise more precisely the position of a gene once linkage has defined the approximate map location. This approach of linkage disequilibrium mapping was recently used to identify the HLA-H gene as the cause of haemochromatosis.56
As discussed previously, it is not currently feasible to search the entire human genome for disease susceptibility genes by association. Case-control studies can be used to examine the role of genes which are thought to be good candidates for disease because of their known biological function. This is worthwhile even if a linkage study has failed to find evidence that the gene is involved in disease susceptibility because of the greater power of association studies. Some examples of the many genes which could be involved in the development of tuberculosis are listed in table 1. One of the major limitations of the candidate gene approach is that there are so many possible candidates that to type them all could involve as much work as an entire linkage based genome screen and still the major disease susceptibility genes could be missed. This is obviously the case if the important genes are not yet cloned or are overlooked, but will also occur if the gene variant typed is not in linkage disequilibrium with the disease causing mutation, which can occur even if the two polymorphisms are within the same gene.
Some candidate genes for susceptibility to tuberculosis
Association studies can produce false positive results when poor matching between case and control groups produces population stratification. This can arise due to unrecognised ethnic admixture and is likely to be a particular problem in Western urban populations where ethnic groups are often defined in very crude terms, such as caucasian, black or oriental. This problem can be overcome by using family based association studies such as the transmission disequilibrium test (TDT)57-59 and haplotype relative risk (HRR) methods.60 ,61 These methods require the genotyping of affected individuals and their parents. Cases are easier to recruit than families with multiple affected individuals, but they can be more difficult to recruit than ordinary cases and controls, particularly in diseases of adult onset where the parents are often unavailable. The TDT compares the frequency with which each allele is transmitted from a heterozygous parent to an offspring with disease. If this is significantly more than the expected 50%, then the allele is either a disease susceptibility allele itself or is in linkage disequilibrium with one. In fig 1 we see that allele 1 has been passed to five out of six possible children from the heterozygous father. If this result was to be replicated in a much larger sample size it would indicate that allele 1 was associated with the disease. The HRR is a similar method to the TDT but compares transmission of genotypes rather than alleles. In fig 1 the 13 genotype has been inherited by four of the six offspring when by chance we would expect it to be inherited by only one child in four. If this result was replicated in a larger sample size it would suggest that the genotype 13 was associated with disease. These tests require both association and linkage to be present to produce evidence of preferential transmission of a disease associated allele and are robust to the effects of population stratification. It is possible to have significant evidence of association on the TDT and/or HRR tests without any evidence of linkage because the association tests are more powerful. Copeman et al found evidence of transmission disequilibrium of chromosome 2q microsatellites in type 1 diabetes for which there was no evidence of linkage.62
Conclusions and future possibilities
There is strong evidence that host genes influence individual susceptibility to tuberculosis. Thus far, however, the identity of the genes involved has remained elusive. Complementary strategies to find these genes include a genome wide linkage screen and association based candidate gene studies. Although candidate gene studies may identify the actual gene(s) involved in the development of tuberculosis, linkage will simply identify the chromosomal location of the genes. The work involved in moving from the mapping of a gene to identifying the gene itself is considerable but will become easier as the Human Genome Project produces more high resolution genetic and physical maps. The identification of tuberculosis susceptibility genes will be greatly facilitated by the genome wide map of expressed sequence tags (EST) which is currently being developed63 ,64 (an EST is a short sequence of coding DNA for which a PCR assay is available). A positional candidate approach could be used to screen systematically all the ESTs from the region of interest to identify the gene(s) responsible for the disease phenotype. Hopefully this approach will eventually enable us to understand what is different about the 10% of the infected population who subsequently develop tuberculosis and enable us to develop new strategies to combat this disease.
Acknowledgments
I am grateful to Professor A V S Hill for helpful discussions and for proofreading this manuscript.
Note
Variation in the NRAMPI gene has now been found to be associated with tuberculosis in West Africans (Bellamyet al. N Engl J Med 1998;338: 640–4).