Article Text

Abstract
BACKGROUND Nitric oxide (NO) may be bronchoprotective in asthma, possibly due to a direct action on airway smooth muscle or through mast cell stabilisation. To investigate this the effects of two doses of nebulised NG-nitro-l-arginine methyl ester (l-NAME), a non-selective NO synthase (NOS) inhibitor, on exhaled NO levels and airway responsiveness to histamine, a direct smooth muscle spasmogen, and adenosine-5′-monophosphate (AMP), an indirect spasmogen which activates mast cells, were evaluated in patients with mild asthma.
METHODS The study consisted of two phases each with a double blind, randomised, crossover design. In phase 1, 15 subjects inhaled either l-NAME 54 mg or 0.9% saline 30 minutes before histamine challenge. Nine of these subjects were studied in a similar fashion but were also challenged with AMP. In phase 2, 13 subjects (eight from phase 1) performed the same protocol but inhaledl-NAME in a dose of 170 mg or 0.9% saline before being challenged with histamine and AMP.
RESULTS The mean (95% CI) reduction in exhaled NO levels after l-NAME 54 mg was 78% (66 to 90) but this did not alter airway responsiveness; the geometric mean (SE) concentration provoking a fall of 20% or more in forced expiratory volume in one second (PC20) after l-NAME and saline was 0.59 (1.26) and 0.81 (1.26) mg/ml, respectively, for histamine and 20.2 (1.7) and 17.2 (1.6) mg/ml, respectively, for AMP. In contrast,l-NAME 170 mg reduced NO levels to a similar extent (81% (95% CI 76 to 87)) but increased airway responsiveness by approximately one doubling dose to both spasmogens; the geometric mean (SE) PC20 for histamine after l-NAME 170 mg and saline was 0.82 (1.29) and 1.78 (1.19) mg/ml, respectively (p<0.001), and for AMP was 11.8 (1.5) and 24.3 (1.4) mg/ml, respectively (p<0.001).
CONCLUSIONS These results suggest thatl-NAME increases airway responsiveness in asthma. This may occur through mechanisms separate from NO inhibition or through pathways independent of those responsible for production of NO measured in exhaled air.
- nitric oxide
- asthma
- airway responsiveness
Statistics from Altmetric.com
Increased airway responsiveness, a key feature of asthma, is characterised by an exaggerated bronchoconstrictor response to a variety of direct and indirect stimuli. The processes underlying increased airway responsiveness are complex and endogenous mechanisms may also exist to protect against theses processes.1Ricciardolo et al recently reported that airway derived nitric oxide (NO) may be important in this respect, suggesting that NO is bronchoprotective in asthma.2
NO is a highly reactive radical formed from the semi-essential amino acid l-arginine by the action of the enzyme NO synthase (NOS).3 It plays a key role in the physiological regulation of the airways and is implicated in the pathophysiology of asthma and other diseases of the airway.4 NO is synthesised by three isoforms of NOS.5 The constitutive isoforms (cNOS) are expressed in neurones (nNOS, type I) and endothelial cells (eNOS, type III) of the airway.6 The third isoform is induced following exposure to proinflammatory cytokines (iNOS, type II) and is expressed in epithelial cells and inflammatory cells of the airway.7 NO can be measured in exhaled air and is increased in asthmatic patients.8
The mechanisms by which NO may be bronchoprotective are not clearly understood, but neuronally derived NO may be important. NO is a neurotransmitter released by inhibitory non-adrenergic non-cholinergic (iNANC) nerves9 and counteracts cholinergic bronchoconstriction.10 In the airways NO activates soluble guanylyl cyclase in target cells, resulting in an increase in cyclic 3′,5′-monophosphate (cGMP) and smooth muscle relaxation.11Inhalation of high concentrations of NO produces bronchodilatation in asthmatic patients12 and inhibition of its production with NOS inhibitors increases airway responsiveness in experimental animals13 ,14 and asthmatic patients.2 In addition to its direct action on airway smooth muscle, NO may also stabilise mast cell activity,15 ,16 a property that may be important since these cells are involved in the pathogenesis of asthma and airway responsiveness to a variety of stimuli.17-19
To investigate further the bronchoprotective mechanisms of NO in asthma and to determine whether these mechanisms reflect inhibition of mast cell function or a direct action on smooth muscle, we studied the effect of nebulised NG-nitro-l-arginine methyl ester (l-NAME), a potent non-selective NOS inhibitor, on exhaled NO levels and airway responsiveness to histamine and adenosine-5′-monophosphate (AMP) in patients with mild asthma. We chose these agents as histamine is a direct smooth muscle spasmogen while AMP exerts its effect indirectly through the release of histamine and other preformed mediators from mast cells.18 We hypothesised that, if NO stabilised mast cell activity in vivo, inhibition of its production would increase airway responsiveness to AMP to a greater extent than histamine.
The study was designed to investigate the effects of a low dose (54 mg) and a high dose (170 mg) of nebulised l-NAME on airway responsiveness. As we had previously shown that l-NAME 54 mg reduced exhaled NO levels by approximately 70% in patients with mild asthma,20 we felt it possible that residual NO may be sufficient to maintain a bronchoprotective action. We anticipated that a higher dose of l-NAME would produce a greater reduction in exhaled NO levels.
Methods
PATIENTS
Twenty non-smoking men (table 1) took part in the study. All patients had mild asthma defined according to American Thoracic Society criteria21 with a forced expiratory volume in one second (FEV1) greater than 70% of predicted and demonstrated a positive skin test in response to common airborne allergens (Dermatophagoides pteronyssinus, mixed grass pollen, or cat fur). None had had an exacerbation of asthma or a respiratory tract infection in the preceding six weeks. Patients took no regular medications for their asthma apart from treating occasional symptoms with intermittent short acting β2 agonists. Patients were recruited through local advertisements and outpatients clinics. Written informed consent was obtained from each patient and the study was approved by the ethics committee of the Royal Brompton Hospital.
Patient characteristics
STUDY PROTOCOL
The study was carried out in two phases according to the dose of nebulised l-NAME under investigation. In phase 1 patients inhaled l-NAME 54 mg and in phase 2, which was commenced only on completion of phase 1, a dose of 170 mg l-NAME was inhaled. Each phase consisted of two parts whereby patients were assessed for the effect of l-NAME on airway responsiveness to histamine and AMP. For practical reasons patients were not required to participate in both phases or parts of each phase, accounting for the different number of patients in phases 1 and 2. However, eight patients did participate in both phases reflecting their availability rather than their response to phase 1.
Phase 1
Fifteen patients were assessed for the effect ofl-NAME 54 mg on airway responsiveness to histamine and nine of these were also assessed for the effect of l-NAME 54 mg on airway responsiveness to AMP. For each spasmogen patients attended the laboratory in the morning on two occasions three to seven days apart. Those patients assessed for both spasmogens therefore attended the laboratory on four occasions and the sequence of challenges was randomised in balanced order such that one challenge agent was administered on the first two visits and the other on the second two visits. After baseline lung function, blood pressure, heart rate, and exhaled NO measurements were performed, patients inhaled nebulisedl-NAME (54 mg in 4 ml 0.9% saline) or placebo (4 ml 0.9% saline) in a double blind randomised order. Thirty minutes after inhalation exhaled NO levels, blood pressure, and pulse rate were measured immediately followed by bronchial challenge.
Phase 2
Thirteen patients, eight of whom had participated in phase 1, underwent the same experimental protocol as phase 1 but inhaledl-NAME 170 mg (or placebo) prior to bronchial challenge. Twelve patients were assessed for the effect of l-NAME 170 mg on airway responsiveness to histamine and AMP, and one patient to AMP alone.
Both doses of l-NAME were administered to eight patients prior to histamine challenge and five patients prior to AMP challenge.
EXHALED NITRIC OXIDE
Exhaled NO was measured using a chemiluminescence analyser (LR 2000, Logan Research, Rochester, UK) sensitive to NO from one to 1000 parts per billion (ppb, by volume). Subjects wore a nose clip and exhaled slowly from total lung capacity (TLC) over 20–30 seconds against resistance provided by a mouthpiece and a wide bore Teflon tube connected to the analyser. NO was sampled continuously at a rate of 250 ml/min from a side arm attached to the mouthpiece. A display unit provided visual guidance for the subject to maintain pressure and exhalation flow rate within the range 3 (0.4) mm Hg and 6 (0.09) l/min, respectively. Results of the exhalation analysis were displayed graphically on a plot of NO and CO2 concentrations, pressure and flow against time. Three successive recordings of end-exhaled NO levels were made and the mean value used in the analysis. The analyser was calibrated daily using NO-free certified compressed air to set absolute zero and then a certified concentration of NO in nitrogen of 90 ppb and 500 ppb (BOC Special Gases, Guildford, UK) and certified 5% CO2 (BOC). The ambient air NO level was recorded and the absolute zero adjusted prior to all measurements. All NO measurements were performed blind by an independent observer who took no further part in the study.
HISTAMINE AND AMP CHALLENGES
Fresh solutions of histamine and AMP (Sigma, Poole, UK) were made up in 0.9% saline in a range of concentrations from 0.0625 mg/ml to 32 mg/ml for histamine and from 0.39 mg/ml to 800 mg/ml for AMP. Each solution was administered from a jet nebuliser attached to a breath activated dosimeter (Mefar, Brescia, Italy). The nebuliser delivers particles with an aerodynamic mass median diameter of 3.5–4.0 μm at an output of 9 μl per breath.
Pulmonary function was assessed by measurement of FEV1 with a dry wedge spirometer (Vitalograph, Buckingham, UK). A standard challenge protocol was used for all provocation tests. All bronchodilators were withheld for at least eight hours before each challenge. Three measurements of FEV1 were taken at one minute intervals, the best of which was taken as the baseline. The subjects then inhaled a series of five breaths of saline as control, followed by a series of five breaths of doubling concentrations of histamine or AMP at three minute intervals, starting with the lowest concentration of each spasmogen—that is, during each sequential inhalation the concentration of histamine or AMP administered was doubled. FEV1 was measured 90 and 150 seconds after each inhalation and the highest value recorded for analysis. The challenges were terminated when a 20% decrease in FEV1 from the post-saline value was recorded. A log dose-response curve was constructed for each agonist and the PC20 calculated by linear interpolation.
NEBULISED L-NAME
Solutions of l-NAME were made up fresh on each study day. l-NAME and placebo were aerosolised by a jet nebuliser (Model CR60, Medic-Aid, Sussex, UK) and inhaled through a mouthpiece with a nose clip worn throughout inhalation. Each solution was inhaled by tidal breathing over 12 minutes.
BLOOD PRESSURE AND HEART RATE
Systolic and diastolic blood pressure and heart rate were measured by an Accitorr 1A monitor (Datascope Corp, New Jersey, USA).
STATISTICAL ANALYSIS
All results were expressed as means with 95% confidence intervals unless otherwise stated. PC20 values were log transformed for analysis and the geometric means calculated. The effect ofl-NAME on responses to provocation of each challenge agent was calculated by comparing the difference in PC20 after inhaling l-NAME and placebo in each subject. This effect was expressed in terms of doubling doses using the formula:
(log10 PC20 l-NAME—log10 PC20placebo)/log10 2
Serial measurements within each phase were compared by repeated measures analysis of variance (ANOVA) followed by pairwise comparisons using the Bonferroni t test. Paired data within each phase and unpaired data between phases 1 and 2 were compared using the appropriate two tailed t test. Statistical significance was taken as p<0.05.
Results
Demographic data did not differ between patients in either phase of the study (table 1). In both phases baseline FEV1 values did not differ significantly between study days (data not given).
PHASE 1: EFFECT OF L-NAME 54 MG ON AIRWAY RESPONSIVENESS TO HISTAMINE AND AMP
Thirty minutes after inhalation l-NAME 54 mg significantly reduced exhaled NO levels by 78% (95% confidence intervals (CI) 66 to 90), representing 79% (95% CI 63 to 96) (p<0.001) in patients undergoing histamine challenge and 74% (95% CI 55 to 95) (p<0.005) in patients undergoing AMP challenge (table 2). In contrast, exhaled NO levels did not significantly change following saline inhalation prior to challenge with histamine (4% (95% CI –20 to 29)) or AMP (4% (95% CI –20 to 28)) (table 2). l-NAME 54 mg did not significantly alter FEV1 (table 3), pulse, or blood pressure 30 minutes after inhalation (data not given).
Individual exhaled nitric oxide levels before (Pre) and 30 minutes after (Post) inhalation of nebulised saline andl-name during each phase of the study
Effect of l-NAME and saline on FEV1 measured at baseline and 30 minutes after nebulisation
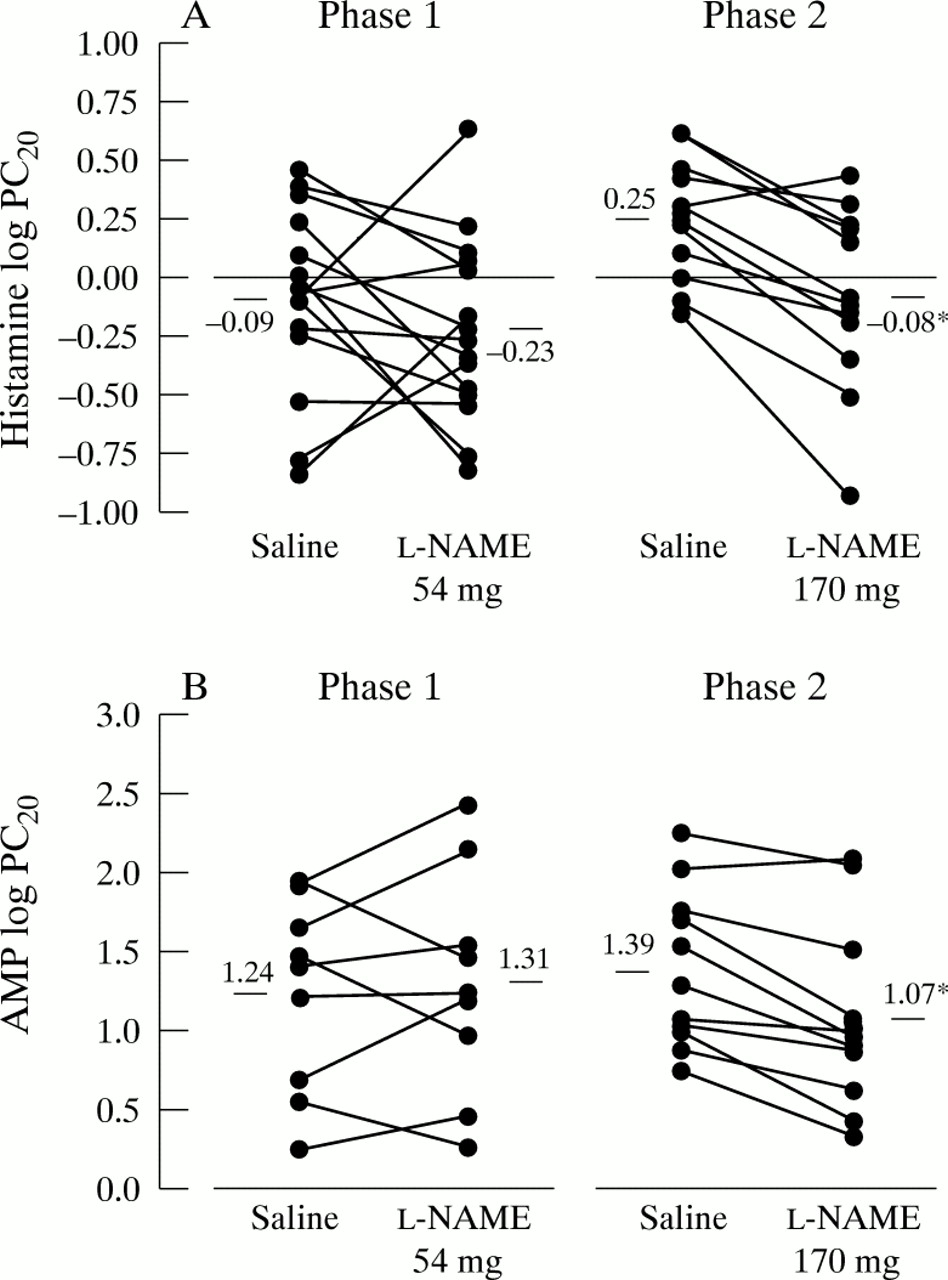
l-NAME 54 mg did not alter airway responsiveness to either histamine or AMP; the mean log PC20 for histamine afterl-NAME was –0.23 (95% CI –0.45 to –0.02) (geometric mean 0.59 mg/ml) and –0.09 (95% CI –0.31 to 0.12) (geometric mean 0.81 mg/ml) after saline (not significant, table 4 and fig 1). The mean log PC20 for AMP after l-NAME was 1.31 (95% CI 0.76 to 1.80) (geometric mean 20.2 mg/ml) and 1.24 (95% CI 0.77 to 1.70) (geometric mean 17.2 mg/ml) after saline (not significant, table4 and fig 1). Thus, relative to saline, l-NAME 54 mg increased airway responsiveness to histamine by 0.5 (95% CI –0.3 to 1.3) doubling doses and decreased airway responsiveness to AMP by 0.2 (95% CI –0.8 to 1.2) doubling doses.
Effect of l-NAME and saline on airway responsiveness to histamine and AMP in each study phase
{kind=link}
Individual effect of l-NAME and saline on airway responsiveness in phases 1 and 2: (A) histamine log PC20, (B) AMP log PC20. Horizontal line represents mean value, *p<0.001 versus saline.
PHASE 2: EFFECT OF L-NAME 170 MG ON AIRWAY RESPONSIVENESS TO HISTAMINE AND AMP
Two patients (nos 4 and 5) did not achieve a 20% fall in FEV1 following the final concentration of AMP at either visit. No data are included on these two patients for these visits only.
Thirty minutes after inhalation l-NAME 170 mg significantly reduced exhaled NO levels by 81% (95% CI 76 to 87), representing 85% (95% CI 77 to 92) (p<0.0001) prior to histamine challenge and 78% (95% CI 69 to 86) (p<0.001) prior to AMP challenge (table 2). Saline inhalation did not significantly alter exhaled NO levels prior to challenge with histamine (2% (95% CI –8 to 4)) or AMP (4% (95% CI –21 to 29)) (table 2). l-NAME 170 mg did not reduce exhaled NO to any greater extent than 54 mg or produce any significant alteration in FEV1 (table 3), pulse, or blood pressure (data not given).
l-NAME 170 mg significantly increased airway responsiveness to both histamine and AMP; the mean log PC20 for histamine after l-NAME 170 mg was –0.08 (95% CI –0.33 to 0.16) (geometric mean 0.82 mg/ml), increasing to 0.25 (95% CI 0.09 to 0.41) (geometric mean 1.78 mg/ml) after saline (p<0.001, table 4 and fig 1). The mean log PC20 for AMP after l-NAME 170 mg was 1.07 (95% CI 0.68 to 1.46) (geometric mean 11.79 mg/ml), increasing to 1.39 (95% CI 1.05 to 1.72) (geometric mean 24.28 mg/ml) after saline (p<0.001, table 4 and fig 1). Thus, l-NAME 170 mg increased airway responsiveness to histamine and AMP by 1.1 (95% CI 0.6 to 1.6) and 1.0 (95% CI 0.5 to 1.5) doubling doses, respectively.
There was no correlation between the degree of reduction in exhaled NO levels and increase in airway responsiveness to histamine (Spearman coefficient r = –0.17, NS) and AMP (r = 0.28, NS).
PATIENTS PARTICIPATING IN PHASES 1 AND 2
Eight patients participating in phases 1 and 2 inhaled both doses of l-NAME prior to histamine challenge. In this subgroup exhaled NO levels were reduced from 22 (95% CI 11 to32) ppb to 4 (95% CI 0 to 10) ppb after l-NAME 54 mg (p<0.01), and from 17 (95% CI 9 to 25) ppb to 3 (95% CI 0 to 6) ppb afterl-NAME 170 mg (p<0.005). l-NAME 54 mg did not significantly alter airway responsiveness to histamine; the mean log PC20 was –0.06 (95% CI 0.43 to 0.32) (geometric mean 0.88 mg/ml) after l-NAME 54 mg and 0.06 (95% CI –0.29 to 0.41) (geometric mean 1.14 mg/ml) after saline. In contrast,l-NAME 170 mg increased airway responsiveness to histamine in this subgroup; the mean log PC20 was 0.0 (95% CI –0.28 to 0.28) (geometric mean 1.00 mg/ml) after l-NAME 170 mg, increasing to 0.28 (95% CI 0.09 to 0.47) (geometric mean l.91 mg/ml) after saline (p<0.01), which corresponds to an increase in airway responsiveness of 0.9 (95% CI 0.3 to 1.5) doubling doses.
As there were only sufficient data on three patients inhaling both doses of l-NAME prior to AMP challenge, no analysis of these data was made.
Discussion
Our results show that inhalation of l-NAME, a non-specific inhibitor of NOS, increases airway responsiveness by one doubling dose to both the direct spasmogen histamine and the indirect spasmogen AMP. Furthermore, in those patients participating in both phases of the study, the action of l-NAME on airway responsiveness to histamine appears to be dose dependent, there being no alteration in PC20 following l-NAME 54 mg, but a reduction of approximately one doubling dose of histamine following l-NAME 170 mg.
There are inevitable limitations to a study of this design. With the relatively small numbers of patients studied it is possible that the power of the study might not have been sufficient to detect a small change in airway responsiveness to the lower dose ofl-NAME, or indeed the observed effect of the higher dose ofl-NAME may have occurred by chance. Our sample size was based on those of Ricciardolo et al 2 who demonstrated statistically significant alterations in airway responsiveness to bradykinin and methacholine with 10 and six patients, respectively, using a similar challenge method. Although there are no published data on the within subject reproducibility of our challenge procedure, which is a modification of the method of Chai et al,22 it is generally accepted that alterations of one doubling dose are within the variability of the method. With this in mind, it is possible therefore that neither dose of nebulisedl-NAME had any true effect on airway responsiveness and the observed effects of both doses were within the reproducibility of the challenge procedure. Looking at individual data it is clear that any effect of l-NAME 54 mg was very variable between subjects, increasing airway responsiveness in some patients but decreasing it in others, which suggests either that, at this lower dose, patients can be characterised into responders and non-responders or, more likely, thatl-NAME had no effect and the observed differences were in fact within the reproducibility of the challenge procedure. In phase 2 of the study, however, the higher dose of l-NAME increased airway responsiveness in 10 out of 12 patients to histamine and nine out of 11 patients to AMP. Despite being within the inherent variability of the challenge, this one doubling dose increase was highly significant suggesting a true effect of l-NAME. Indeed, post hoc power analysis showed that 12 patients were sufficient to demonstrate a one doubling dose shift in PC20 with 90% power following l-NAME 170 mg. Furthermore, in the eight patients who inhaled both doses of l-NAME prior to histamine challenge the variability of response to the lower dose was much greater than the higher dose, supporting the observations of the study as a whole.
These results concur with the findings of Ricciardolo et al 2 and suggest that endogenous NO is bronchoprotective in asthma. Interestingly, Ricciardolo et al did not measure exhaled NO levels in their study and we have demonstrated that, whilst l-NAME 170 mg increased airway responsiveness to both spasmogens, it did not reduce exhaled NO levels to any greater extent than l-NAME 54 mg which had no effect on airway responsiveness. Furthermore, we did not find any correlation between the degree of reduction in exhaled NO levels and increases in airway responsiveness after the higher dose of l-NAME. This therefore implies that NO—or, at least, NO measured in exhaled air—does not play a critical role in bronchoprotection and suggests that l-NAME is exerting an additional, as yet unknown, effect on the mechanisms of airway responsiveness separate from NOS inhibition. Whilst both doses of l-NAME are of the same pH, the higher dose has greater tonicity and this may account for the observed differences in our study. Although speculative, an alternative explanation is that maximal suppression of exhaled NO has occurred with the lower dose of l-NAME, but higher doses are able to penetrate further into the airway and inhibit NOS in cells that are not responsible for the generation of NO measurable in exhaled air. The cellular source of exhaled NO is not precisely known. iNOS expression is increased in airway epithelial cells in asthmatic subjects, reflecting underlying inflammation,23 ,7 and it is likely that the increased levels of NO observed in asthma are derived from this source. l-NAME at both doses may be able to inhibit constitutive and inducible NOS within the epithelial layer and it is possible that this layer may act as a metabolic or diffusional barrier for l-NAME, preventing the lower dose in particular from acting on sources of NOS situated more deeply within the airway parenchyma. Ricciardolo et al demonstrated that inhalation of nebulised l-NMMA l mg increased airway responsiveness to both bradykinin and methacholine in asthmatic patients.2Based on the assumption that l-NMMA exerted its effect through inhibition of airway NO, they concluded that NO was bronchoprotective in asthma despite not measuring exhaled NO levels in their study. In the knowledge of the results of our study we believe that this conclusion is less secure and, in a similar manner,l-NMMA may have exerted its effect through mechanisms separate from NO inhibition. Interestingly, the dose ofl-NMMA was substantially less than the lower dose ofl-NAME in our study which had no effect of airway responsiveness but was administered through a face mask over 30 minutes. In contrast, we administered l-NAME through a mouthpiece over 12 minutes with a nose clip worn throughout. The greater exposure to l-NMMA over this time course is likely to account for the differences in dose effects observed between the two studies.
If l-NAME at the higher dose does inhibit NO not measurable in exhaled air, the mechanisms through which NO exerts its bronchoprotective effect are unclear. In the study by Ricciardoloet al the potentiation of bradykinin induced bronchoconstriction (3.6 doubling doses) was greater than methacholine induced bronchoconstriction (1.3 doubling doses).2 Indeed, the degree of potentiation of airway responsiveness to methacholine was similar to that for histamine and AMP in our study. They suggested that this difference in potentiation may reflect the inhibition of NO released by the action of bradykinin on neural and non-neural cell types in the airways. It is therefore possible that bradykinin exerts a greater effect on NO release than other spasmogens. Inhibition of neuronal NO synthesised constitutively by iNANC neurones may be important in this respect. In human airways in vitro l-NAME has no effect on airway smooth muscle resting tone but enhances cholinergic neural constrictor responses to electric field stimulation.10 This suggests that there is no basal release of NO from iNANC neurones and that stimulation evokes release of endogenous NO that acts to inhibit cholinergic contractile responses. Both AMP24 and histamine25 exert additional reflex vagal bronchoconstriction through stimulation of airway sensory nerves. Such stimulation may also activate iNANC pathways evoking NO release. Inhibition of NO synthesis therefore might be expected to attenuate the iNANC response and enhance bronchoconstriction. Indeed, the findings of our study are in keeping with these observations as l-NAME did not affect airway resting tone, with no alteration in FEV1 30 minutes after inhalation, but did increase airway responsiveness to both spasmogens. Furthermore, as iNANC neurones are situated more deeply within the airway parenchyma it is tempting to speculate that only the higher dose of l-NAME is able to inhibit NOS within these neurones.
In experimental animals there is considerable evidence to suggest that endogenous NO regulates the reactivity of mast cells.15 We hypothesised that, if NO exerted mast cell stabilising properties in vivo, inhibition of its production would increase airway responsiveness to the indirect spasmogen AMP to a greater extent than the direct spasmogen histamine, as has been observed for β2agonists.26 In our study airway responsiveness to both agents was increased to the same extent, suggesting that the potential mast cell stabilising properties of endogenous NO may not be functionally important in human airways. It is possible that NO acts principally through functional antagonism at the level of the airway smooth muscle. However, it is also possible that there is a substantial difference in the availability of NOS and NO to l-NAME within mast cells, iNANC neurones and bronchial epithelium, and thus inhibition of mast cell NOS may not have been sufficient to significantly attenuate NO regulating mast cell reactivity.
The differential effects of 54 mg and 170 mg of nebulisedl-NAME are important for the planning of future studies investigating NOS inhibitors and the role of endogenous NO in asthma. If the lack of effect of lower doses of l-NAME does reflect poor penetration through the epithelial layer, synthesis of physiologically important endogenous NO within deeper structures may not be inhibited. This may lead to the misinterpretation of negative studies. l-NAME 170 mg was well tolerated without adverse effects or alteration in blood pressure or heart rate and we believe this dose should be used in future studies.
In conclusion, this study has attempted to evaluate further the role of endogenous NO in the regulation of airway smooth muscle. We cannot conclude definitively that NO is bronchoprotective in asthma as it appears that nebulised l-NAME exerts additional effects over and above NO inhibition. Further studies need to be undertaken to determine the exact mechanisms by which l-NAME attenuates airway responsiveness in human airways as well as defining its site of action. The effects of nebulised NOS inhibitors at higher doses should also be considered and other indirect bronchial challenges investigated.
Acknowledgments
Supported by a grant from Imperial College School of Medicine.