Article Text

Statistics from Altmetric.com
Since the isolation of the gene responsible for cystic fibrosis in 1989 we have witnessed rapid progress in our understanding of this common genetic disorder. Progressively more detailed knowledge of the disease causing mutations, the gene expression patterns, the protein structure, and its function as an ion channel has allowed the development of reliable screening programmes, the creation of animal models, and the design of novel therapeutic approaches. The biological basis of many aspects of the disease phenotype are becoming clearer, including recent significant advances in understanding the pathogenesis of the characteristic and ultimately fatal lung disease. While this great expansion of knowledge has started to bridge the gap between our understanding of the genetic defect and the well documented clinical sequelae, it has also served to demonstrate the mechanistic complexity of this “simple” single gene defect.
Cystic fibrosis and the cloning of the gene
Cystic fibrosis is the most common lethal autosomal recessive genetic disorder in Caucasian populations with a carrier frequency of one in 25. It affects about one in 2500 live births and has a median life expectancy of 28 years.1
The characteristic manifestations are due primarily to dysfunction of exocrine glands, producing viscid, dehydrated secretions. The resultant phenotype is of salty sweat, pancreatic insufficiency (with consequent malabsorption and failure to thrive), intestinal obstruction (with meconium ileus in 10–20% of patients in the first few days of life), infertility and severe pulmonary disease. The electrophysiological profile of affected tissues shows characteristic abnormalities in the transport of chloride and sodium ions across the apical membrane.2
The recent advancement of our knowledge was sparked by the cloning, in 1989, of the gene responsible for the disease—the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Located on the long arm of human chromosome 7, it spans over 250 kb, contains 27 exons, and encodes a 1480 residue transmembrane glycoprotein. The protein consists of two repeated motifs made up of six membrane spanning segments, and a nucleotide binding domain (NBD) separated by a regulatory domain (R domain) with multiple potential phosphorylation sites. It has been shown to function as a cAMP regulated chloride ion channel at the apical membrane of epithelial cells.3
The clinical problems in cystic fibrosis reflect the tissue and cell specific expression pattern of CFTR, being highly expressed in the epithelial cells of the sweat ducts, the pancreatic ducts, the digestive tract, particularly in the crypts of the small intestine and Brunner’s glands, the biliary ducts, the salivary glands, the reproductive organs and the lungs, particularly in the serous cells of the submucosal glands and in the respiratory epithelial cells.1
Research into cystic fibrosis was further boosted by the development of transgenic cystic fibrosis mouse models after the murine homologue (Cftr) of the human gene was isolated. The murine CFTR protein is very similar to the human (78% overall sequence identity at the amino acid level), particularly in the two nucleotide binding folds, and the cAMP dependent chloride ion transport properties of wild type mice are similar to those of normal human subjects.4The first mouse models of cystic fibrosis were created within three years of the isolation of the CFTR gene, using gene targeting in embryonic stem cells to disrupt the Cftrgene, and to date 10 variants have been created, all demonstrating the characteristic abnormalities in cAMP mediated chloride ion conductance, but importantly also exhibiting a range of clinical phenotypes.5
CFTR mutations
Since the isolation of the CFTR gene there have been over 600 different mutations identified.6 The most common of these, accounting worldwide for 68% of the mutant alleles, is ΔF508, a three base pair deletion in exon 10 that removes phenylalanine 508 from the first NBD. Only a handful of others are common (>1% of the total), but some are significantly enriched or depleted in specific ethnic populations. The vast majority are very rare, constituting a practical challenge to the clinical geneticist with respect to molecular diagnosis, but also a valuable epidemiological study which links protein structure to function and genotype to phenotype. All classes of mutation are observed, with the highest density of mutations occurring in the two nucleotide binding domains. Most known cystic fibrosis mutations occur in conserved regions suggesting conservation of function across species.
Mutations in CFTR may result in: (1) defective CFTR production, such as R553X, due to unstable mRNA and/or premature protein truncation, (2) defective processing of CFTR, such as ΔF508 or G480C, where the mutant protein is not processed to its mature glycosylated form and is not correctly localised to the apical membrane, but is retained in the endoplasmic reticulum and degraded. However, under permissive conditions in vitro, such as reduced temperature, correct localisation of mature protein can occur where it can function normally (in the case of G480C), or suboptimally (in the case of ΔF508), or (3) defective ion channel function, such as G551D or R117H, in which case some of the mutant protein becomes correctly localised but results in either very little residual function (in the case of G551D) or a substantially reduced level of ion transport (in the case of R117H).
In each class of mutation the level of functional CFTR at the apical membrane of epithelial cells in patients with cystic fibrosis falls below a critical level, resulting in the characteristic clinical abnormalities observed in the organs in which CFTR is expressed.3
The significant advances in our understanding of the disease and the development of new technologies now allows prenatal diagnosis, with diagnostic accuracy of 100% in almost all cases where an affected family member can be examined, and the implementation of carrier screening programmes to help individuals from affected families to make informed decisions in family planning. However, the picture is still complicated in a significant subset of unusual cases.
The genotype-phenotype relationship
Understanding the relationship between genotype and phenotype is obviously important from the perspective of prenatal testing, but also for what it can tell us about the pathophysiology of the disease and prospects for intervention.
The mutations in CFTR are usually classified as either “mild” or “severe” depending upon whether they confer pancreatic sufficiency (PS) or insufficiency (Pl), with one “mild” allele being enough to result in pancreatic sufficiency. However, this relatively clear relationship between genotype and phenotype does not apply to all the affected systems.
The common ΔF508 mutation is classified as “severe”, conferring pancreatic insufficiency and associated with meconium ileus in approximately 10% of affected neonates. The G551D mutation, although also “severe” and conferring pancreatic insufficiency, is observed to be associated with a much lower incidence of meconium ileus, while the R117H mutation is a “mild” mutation in which pancreatic sufficiency is generally observed. However, all three mutations are associated with infertility and lung disease.7
The quantity of normal functional CFTR can be affected by alternative splicing of mRNA such as that associated with the 5-thymidine (5T) variant of the intron 8 polypyrimidine tract (IVS8-T).8This variant gives rise to two transcripts—one normal and one with an in frame deletion of exon 9, the protein product of which is devoid of cAMP activated chloride conductance activity. The level of normal transcript has been shown to be dependent upon the associated IVS8-T (with the 5T variant associated with the lowest levels, increased with the 7T variant and greatest with the 9T variant), to be tissue dependent, and consequently to affect phenotype.9
Asymptomatic individuals homozygous for the 5T allele and expressing as little as 8% of normal CFTR in some tissues have been documented,8 while in other instances these individuals present with congenital bilateral absence of the vas deferens (CBAVD). Other individuals with CBAVD have one recognisable CFTRmutation and the 5T variant on the other chromosome. Furthermore, individuals carrying the R117H allele are typically more severely affected if the R117H mutation is in cis with the 5T polymorphism as opposed to the 7T variant, the associated IVS8-T affecting the amount of already functionally impaired CFTR that is correctly spliced to influence the phenotype.7
Thus, detailed studies of the pathophysiological consequences of different mutations and splicing abnormalities have provided some insight into the effects of specific alterations in the quality or quantity of CFTR and it has been possible to correlate the level of CFTR function with phenotype in an organ specific manner. In this way we can examine the variable sensitivity of different tissues to CFTR dysfunction and address the important question of how much functional CFTR is required for therapeutic benefit.
In studies using cystic fibrosis epithelial cell monolayers the results have suggested that functional CFTR is only required in approximately 10% of cells to restore the bioelectric phenotype to normal.10 Further work with transgenic cystic fibrosis mice broadly supports these observation in vivo and demonstrates a non-linear relationship between gene activity and intestinal phenotype, the system that is most severely affected in the mouse models.11 The specific pathology observed and the degree of severity varies considerably between models, resulting in marked differences in survival rates from approximately 10% in thecftrm1UNC absolute nulls (in which no CFTR is produced) to approximately 90% in thecftrm1HGU mice (which produce about 10% normal CFTR). The results indicate that quite small corrections in gene activity can have marked electrophysiological and dramatic pathological consequences. Together with clinical observations of diminished disease severity in the presence of residual function alleles, the non-linear relationship between genotype and phenotype underpins the hopes for successful treatment by gene therapy.
Phenotype modification
While this research is shaping the relationship between specific mutations, levels of functional CFTR, and some aspects of phenotype, the picture is incomplete with further significant variability being introduced as a result of independently segregating modifier genes and various environmental factors.
Further insights may come from comparisons of the phenotype of individuals with the same genotype and ethnic background in very different environments and from studies of cystic fibrosis twins with age matched siblings in similar environments and with the sameCFTR genotype.
The cystic fibrosis mouse models will also be a valuable resource in this regard, not least because of the ability to control both the environment and the genetic cross to compare the effect of variousCftr mutations on different inbred strain backgrounds. In this manner a locus has already been identified on the proximal region of mouse chromosome 7 that modifies the onset and severity of intestinal pathology and survival.12 This region shows conserved synteny with human chromosome 19q13. It has been suggested that this modifier could take the form of a calcium dependent chloride channel that may be able to compensate partially for the absence of CFTR and alleviate the symptoms.
A significant degree of variability has also been observed betweencftrm1HGU mice on different inbred strain backgrounds with regard to airway electrophysiology and the response to inhaled pathogens.13 Studies are underway to isolate these modifier genes.
The ability of other genes to modify the progression and severity of cystic fibrosis through alternative ion channels, the regulation of normal CFTR production or other mechanisms is likely to explain some of the variability but it is probable that environmental influences also play an important role in humans, particularly in the development of lung disease.
Other roles for CFTR
Although the primary defect in cystic fibrosis is in chloride transport, the other classical abnormality observed in cystic fibrosis cells is a marked increase in the level of sodium absorption by the amiloride sensitive epithelial sodium channel (ENaC) across the apical membrane of the epithelial cells. It has been shown in vitro that ENaC activity is decreased in cells overexpressing CFTR, and that cAMP has an inhibitory effect on ENaC activity in the presence of CFTR, but a stimulatory effect in its absence.14
CFTR has also been postulated to have a role in the transport of ATP, either directly as an ATP channel or as a regulator of ATP transport across both the apical membrane and the membranes of the intracellular organelles.15
A role in chloride transport has also been suggested for CFTR across the membranes of intracellular organelles. In this manner it might cause a myriad of other effects by affecting the pH balance of the organelles and consequently the processing of other glycoproteins, altering the pattern of sulphation and sialylation.16
These secondary functions of CFTR serve to remind us of the gaps in our knowledge and the complexities of the physiological processes in which CFTR is involved.
Lung disease in cystic fibrosis
While the phenotypic analysis described has provided a considerable amount of information about disease of the vas deferens, pancreas and digestive tract, the severity and course of pulmonary disease are not clearly predicted by the CFTR genotype.
The precise aetiology of lung disease in cystic fibrosis is still relatively poorly understood, but is characterised by chronic microbial colonisation and repeated acute exacerbations of pulmonary infection, with distinctive bacterial flora precipitating progressive irreversible inflammatory lung damage. Excessive mucus production, airway obstruction and atelectasis, cytokine imbalance and a neutrophil dominated cellular response, producing excessive amounts of elastase, end in severe bronchiectasis and respiratory failure.
BACTERIOLOGICAL PROFILE OF THE CYSTIC FIBROSIS LUNG
The environment of the cystic fibrosis lung is unique and results in characteristic disease patterns and a specific spectrum of cystic fibrosis associated lung pathogens. Infants with cystic fibrosis are typically subject to respiratory infections—both with viruses such as respiratory syncytial virus (RSV), and bacteria, such asStaphylococcus aureus and Haemophilus influenzae, with S aureus predominant in the first two years of life.17 Aggressive antibiotic therapy has enabled increased control over infection with these bacteria that once killed the majority of cystic fibrosis patients in infancy.
After the first two years Pseudomonas aeruginosapredominates, with evidence of infection in the lower airways in a third of cystic fibrosis infants by the age of five years.18 Early chronic colonisation with non-mucoid strains may sometimes be eradicated, but strains tend to persist in individuals and alter their phenotype. While there is variability in the time scale for the emergence of alginate producing mucoid variants of P aeruginosa, it can occur in as little as three months after the initial colonisation and the FEV1, the single best predictor of mortality, will then decline more quickly in those patients in whom P aeruginosa has become mucoid. Despite considerable advances in the development of anti-pseudomonal therapy, it remains palliative and seldom results in eradication of mucoid strains.17
In the past decade infection with Burkholderia cepacia has become an increasing concern. Strains of this multi-resistant organism can also colonise the lungs of patients with cystic fibrosis and some strains demonstrate direct cross infection between patients.17 B cepacia is resistant to a wide range of antibacterial agents, including cationic peptides,19 and certain strains are capable of causing rapid clinical deterioration, fulminating pneumonia, bacteraemia, and death.
DEVELOPMENT OF CYSTIC FIBROSIS LUNG DISEASE
The analysis of necropsy material from newborn infants with cystic fibrosis who die in the first few days of life suggests that their lungs are structurally and functionally normal.20 However, mild histological abnormalities have been observed in the respiratory tracts of some cystic fibrosis infants who died of meconium ileus in whom there was no evidence of pulmonary infections,21 and increased levels of IL-8 and neutrophils have been found in bronchoalveolar lavage fluid from infants with cystic fibrosis compared with controls, despite negative cultures for the usual cystic fibrosis associated pathogens.22 This evidence suggests that pulmonary abnormalities may precede full blown chronic airways colonisation, but the mechanisms that precipitate these changes remain uncertain. Whether this represents an exaggerated response to minor infectious stimuli that may themselves have been removed, an inappropriately regulated response or, perhaps, signs of developmental abnormality, it is quite conceivable that this is the beginning of a vicious circle of inflammation that will not properly resolve.
The precise mechanisms by which defective transepithelial chloride ion transport results in the observed pulmonary pathology remain poorly understood. Indeed, it has been suggested that, while CFTRmutations cause primary lung dysfunction, this alteration to the lung environment may be a point of no return after which a self perpetuating process has begun and subsequent pathology need not result from a direct interaction with CFTR. This theory attempts to distinguish between cystic fibrosis disease, caused and maintained by CFTR dysfunction, and cystic fibrosis lung disease, a disease phase triggered by the former,23 and could, if correct, have significant bearing on the direction of future research and the emphasis in the design of novel treatments.
Whether CFTR dysfunction directly results in the creation of an abnormal lung environment which harbours infection, or an abnormal response to cystic fibrosis associated pathogens produces the characteristic pathology and subsequent clinical sequelae, are questions that have been difficult to address. The dissection of the complex pathogenesis of this disease has suffered from the limitations imposed by restricted access to clinical material, a reliance on necropsy specimens with “end stage” lung disease, and in vitro techniques. The development of mouse models, better in vitro techniques, and a re-evaluation of the old model in the light of new discoveries has meant that possible answers to some of the questions are now being revealed.
LUNG DISEASE IN CYSTIC FIBROSIS MICE
The value of the cystic fibrosis mouse models in dissecting the pathogenesis of cystic fibrosis and developing novel treatments derives from the extent to which they mimic the lung disease seen in patients with cystic fibrosis. There is no evidence of pulmonary abnormality at birth in any of the models and, despite defects in the electrophysiological profiles of all the mutant mice, no spontaneous disease has been observed.
In studies of the cftrm1HGU model no lung disease was observed at birth, nor in animals raised in isolators. However, histopathological evidence of pulmonary pathology was noted in mice reared in normal animal house conditions in which they are exposed to a low level of background pathogens. Although no significant difference existed between the genotypes, there was a trend towards worse pathology in the cystic fibrosis mutant mice. Impaired airway clearance of nebulised S aureus and B cepaciaand the development of significantly more severe pathogen-specific lung disease after repeated exposure to these bacteria was seen in the mutant animals in comparison with their non-cystic fibrosis littermate controls.24 The spectrum of abnormal pathology included inflammatory infiltration, goblet cell hyperplasia and metaplasia, mucus retention, bronchiolitis, pneumonia and oedema, indicating a similarity to the histopathology of cystic fibrosis lung disease in humans. These studies suggest an increased predisposition to lung disease in the cystic fibrosis mice when exposed to specific bacteria, and also that this exposure to pathogens is crucial to exacerbate the underlying defect.
Further studies on the cftrm1HGU mice have shown a significant increase in inflammation in the tracheal lamina propria of cystic fibrosis mice and significant impairment of mucociliary transport compared with their littermate controls.25
In recent studies using P aeruginosa in an agar bead infection model cftrm1UNC mutant mice have been shown to have significantly higher mortality rates and raised proinflammatory cytokines prior to death in comparison with wild type littermates.26
Thus, the development of transgenic cystic fibrosis mice has provided the opportunity to start to investigate disease pathogenesis in vivo, to interrupt the process at defined stages and under controlled environmental conditions (using appropriate controls), to make comparisons between different mutations of Cftr, to search for modifier genes and, as a consequence, to begin to dissect the web of initiating and potentiating factors downstream of CFTR that are responsible for the characteristic pulmonary disease.
It is hoped that within the next couple of years a sheep model of cystic fibrosis may be available to supplement the current mouse studies, providing new opportunities that will result from a model pulmonary system with greater anatomical and physiological similarities to man as well as the obvious advantages of size alone.27
MECHANISMS OF THE PATHOGENESIS OF CYSTIC FIBROSIS LUNG DISEASE
The traditional model for cystic fibrosis lung disease involves mucus hypersecretion and dehydration which upsets normal mucociliary clearance mechanisms. Bacterial infection and hyperviscous secretions result in accumulation of mucus in the airways and airflow obstruction. Colonisation with cystic fibrosis associated pathogens occurs and persistent neutrophil influx contributes to an excessive and damaging inflammatory response and cytokine imbalance. Suppuration leads to ulcerative bronchitis and, ultimately, bronchiectasis.
Although studies on mucus from patients with cystic fibrosis suggest a spectrum of rheological abnormalities,28 mucociliary clearance studies have been inconclusive29 ,30 and there is no evidence to support the hypothesis that the abnormal function of CFTR, per se, can induce an impaired mucociliary clearance in the absence of infection. It is also worth noting that classical cystic fibrosis lung disease is not observed in patients with primary ciliary dyskinesia syndrome nor in pseudohypoaldosteronism. The former is a disorder of mucociliary clearance that results in rhinitis and bronchitis and can lead to bronchiectasis, but generally causes a much less severe lung disease than cystic fibrosis.31 The latter is a salt-wasting disorder due to mutations in subunits of ENaC. The systemic form results in increased sweat sodium and chloride levels and chronic excessive secretion from the respiratory tract. However, despite predisposition to pulmonary infections and asthma like symptoms, they seldom develop bronchiectasis.32Furthermore, neither condition displays the characteristic bacteriological profile of cystic fibrosis lung disease.
While evidence continues to show that P aeruginosa can certainly upregulate mucin genes,33 new discoveries have reshaped the original model and started to bridge the gap between the genetic defect and resultant pathology in a more satisfactory manner.
The current theories place greater emphasis upon compromised innate defence mechanisms within the airway surface fluid as a result of ionic imbalance caused by CFTR dysfunction and the interaction between epithelial cells and bacteria.
An innate defence mechanism in the airway surface fluid
The exciting discovery in 1996 that airway surface fluid had salt sensitive antibacterial activity34 has precipitated the discovery of an innate lung defence system comprising a variety of interacting antimicrobial factors including naturally occurring antimicrobial peptides, human β-defensins 1 and 2 (hBD135 and hBD236), lysozyme, and lactoferrin.37
The antibacterial property of airway surface fluid from primary cultures of normal airway epithelial cells are observed to be markedly reduced in the airway surface fluid of cultures of cells from patients with cystic fibrosis. Increasing the salt content of normal airway surface fluid dramatically impairs its antibacterial activity while that of airway surface fluid from patients with cystic fibrosis can be restored by lowering its salt concentration, which suggests that the components of the defence system are intact but are unable to function correctly in an environment with high levels of sodium chloride.34 This led to speculation that CFTR dysfunction alters the ionic environment of the cystic fibrosis lung, impairing the first line of defence against bacteria and viruses, predisposing these patients to infection and possibly precipitating a more pronounced compensatory response from other components of the defence system, affecting neutrophils, macrophages and cytokine production.
This presents an attractive explanation but attempts to measure accurately the concentration of sodium and chloride in the airway surface fluid of individuals with and without cystic fibrosis in vivo have provided conflicting results and been hampered by sampling difficulties. Most recent results suggest that the airway surface fluid from patients with cystic fibrosis does indeed have a higher concentration of sodium chloride,34 ,38 perhaps as a result of a failure of reabsorption similar to that in the sweat gland,39 but other studies have found no significant difference.40 The development of techniques to provide accurate measurements will be crucial for the development of this hypothesis.
A murine homologue of hBD1 (Defb1) has now been reported.41 ,42 Defb1 has been shown to have a similar expression pattern to hBD1, including expression in the airways, and functional studies using a synthetic peptide have shown a salt sensitive antimicrobial activity similar in profile to that of its human counterpart.42 These results suggest that murine airway surface fluid will be a good model for the further study of the naturally occurring peptide antimicrobials and that compromise of the same innate lung defence mechanisms in cystic fibrosis mice may be involved in the abnormal responses observed when exposed to pathogens.
Epithelial cell interaction
The interaction between bacteria and epithelial cells encompasses two areas of research—the adherence of P aeruginosa to asialylated glycoproteins on the cell membrane and the internalisation of P aeruginosa by epithelial cells.
It has been reported that, in vitro, P aeruginosa adheres to respiratory epithelial cells from patients with cystic fibrosis (particularly ΔF508 homozygotes) significantly better than to controls43 and with increased affinity to damaged and regenerating epithelial cells,44 in addition, that P aeruginosa pilin (an adhesin responsible for about 50% ofP aeruginosa binding43) recognise and bind to cell surface asialoganglioside 1 (aGM1), which is present in higher levels in cystic fibrosis airway epithelia than in controls at the expense of the sialylated protein43 (possibly as a consequence of defective acidification of intracellular organelles as a result of CFTR dysfunction).16 The levels of aGM1 on cystic fibrosis cells are also found to be further increased in response to P aeruginosa neuraminidase and are greater on regenerating cells. Further evidence comes from experiments in which adherence of P aeruginosa is found to be reduced in primary culture of respiratory epithelium from patients with cystic fibrosis after transfection with human CFTRgene therapy constructs.45
These data thus suggest putative pathogen specific binding sites on airway epithelial cells with potential for interaction with bacterial exoproducts, present in greater quantity on cystic fibrosis cells and after injury. However, the consequence of these observations and their in vivo relevance are as yet unclear. Furthermore, they conflict with post mortem evidence in which P aeruginosa is observed as microcolonies in an extracellular mucus layer, and rarely adherent to cells.46
An alternative avenue of research has explored the possibility that, as part of the host defence mechanism, airway epithelial cells may internalise P aeruginosa, with the lipopolysaccharide core oligosaccharide acting as the bacterial ligand for ingestion.47 It is suggested that the first extracellular domain of CFTR acts as a specific cellular receptor. Interestingly, a deletion from the N-terminal to the fourth membrane spanning domain has no effect on the ion channel properties of CFTR.48
This process of internalisation has been found, in vitro and in vivo, to be specifically blocked by appropriate antibodies or peptides and, in vitro, to be impaired in cystic fibrosis cells. Preliminary evidence suggests that the low level of CFTR expression on the apical surface of airway epithelial cells may be significantly upregulated by unknown mechanisms upon contact with P aeruginosa, form a point of adherence with the bacteria, and consequently ingest the organism. It is postulated that exposure to low environmental levels of bacteria, focal internalisation and cell shedding (perhaps by a minority of cells ready to be desquamated in a manner similar to that in corneal infection with P aeruginosa) might thus clear these pathogens, a mechanism that would be impaired in the absence of apical CFTR or in the presence of correctly localised but dysfunctional protein.49
However, once again the in vivo relevance of these observations is uncertain. Initial experiments to study the internalisation in cystic fibrosis mutant mice have suggested that strain differences may have a more substantial role than the mutation itself and in an earlier study, using an identical model of mouse infection, internalisation was seen mainly in those animals that developed severe pneumonia and postulated as a route to more widespread infection. In addition, it has yet to be shown that the monoclonal antibodies used in these studies detect CFTR alone and not a previously reported cross reacting protein.49
While the two theories of interaction between epithelial cells andP aeruginosa appear to be contradictory, the accumulating data suggest that interaction may play an important part in the development of lung disease in cystic fibrosis and, particularly, in conferring the organism selectivity observed. However, both hypotheses fail to address the issue of the mucus barrier that protects the cells from the luminal contents and the observation that the disease process originates in the smallest airways.
The new model (fig 1) now proposes that the CFTR dysfunction directly affects the ionic composition of the airway surface fluid and compromises the antimicrobial activity of the salt sensitive first line of lung defence. This breached, the organisms have a better chance of colonising the lung. A compensatory response from other elements of the defence mechanism may occur, with an increased neutrophil influx, the release of damaging elastase, cytokine imbalance and the triggering of inflammatory cascades, while cell-bacteria interactions may select for specific organisms. The continuing presence of these organisms may then further upregulate the expression of mucin producing genes.
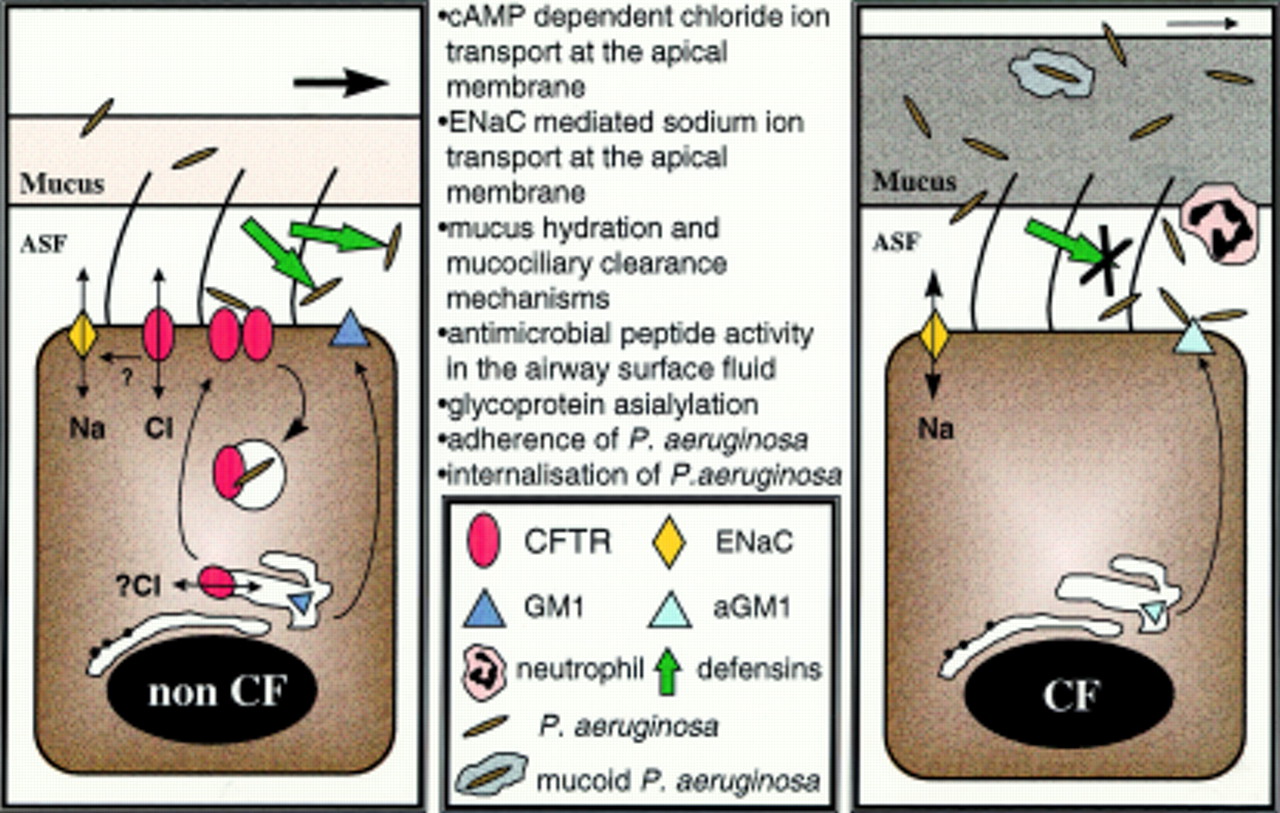
{kind=link}
Effects and influences of CFTR in airway epithelial cells.
This, of course, is only half of the equation in which the abnormal environment must in some way stimulate phenotype alterations in the bacteria, with P aeruginosa undergoing mucoid change, thus enhancing its survival and again interacting with its host to further alter its environment.
The model goes some way to explain the pathogenesis of cystic fibrosis lung disease, but remains incomplete. Development of animal models in which components of the innate lung defence system are knocked out are eagerly awaited. To what extent the absence of this mechanism in isolation mimics cystic fibrosis lung disease will help to test the new model, as will the development of salt insensitive defensins for possible therapeutic use and in vivo studies examining the interaction between epithelial cells and bacteria.
The model fails to address the role of the submucosal glands, but with high levels of CFTR expression in the serous cells and as a major source of fluid and mucin in the lungs it seems possible that they may have a role to play in the maintenance of the ionic composition of the airway surface fluid. A recent study in thecftrm1HGU mouse suggests that submucosal glands may indeed have a role in the development of lung disease and highlights this as an area worthy of further study.50
The current hypothesis also offers no explanation for the susceptibility to pulmonary colonisation with B cepacia, which has been found to be resistant to cationic peptides and specifically to killing by synthetic hBD1.42This observation suggests that the impairment of some component of lung defence other than the antimicrobial properties of airway surface fluid may be responsible. Pulmonary infection with B cepacia is very rare other than in cystic fibrosis, but does occur in chronic granulomatous disease (CGD). This is a condition in which there is failure of oxidative killing by the phagocytic cells as a result of an inability to produce a respiratory burst51 and is characterised by recurrent infections, suppurative granulomas, and infiltration of the viscera with pigmented lipid-laden macrophages. It remains to be seen whether dysfunctional CFTR can also cause a defect in oxidative killing.
Treatment
As our understanding of the molecular and biological basis of cystic fibrosis becomes more comprehensive, so the ultimate goal of successful treatments becomes more accessible. With every new insight into the complexity of the pathogenesis comes a better appreciation of both the limitations of current treatments and of new opportunities for therapeutic intervention (table 1). While it is beyond the scope of this review to detail the current treatment regimes and trials of novel therapies in progress, it is worth considering a few important issues.
Lung disease in cystic fibrosis
Treatment must aim to improve quality of life and, if possible, to provide a cure rather than simply to alleviate symptoms partially. There is a continuing need and justification for the development of new antimicrobial agents, mucolytics, and antiproteases. More directly relevant to the new genetic knowledge is the scope for (a) relocating CFTR to the apical surface, (b) upregulating the function of correctly localised mutant CFTR, (c) overcoming stop mutations at the protein translational level, or (d) compensatory regulation of alternative ion channels.52 ,53 Not surprisingly and, again, appropriately, considerable effort is also being invested in gene therapy to replace the faulty gene directly and thus to correct all the downstream consequences that arise from CFTR dysfunction at once.
Gene therapy for cystic fibrosis has been the victim of initial overenthusiasm in some quarters, perhaps due to the apparent simplicity of the concept. Adenovirus, adeno-associated virus, and cationic liposomes have each been tested in phase I clinical research studies for transfer of the CFTR gene to respiratory epithelium.54 When proper account is taken of the highly experimental nature and early stage of this research, the results, in terms of both safety and efficacy, have been reasonably encouraging. The hurdles which still need to be overcome are, however, clear for all to see (table 2) and this has given rise to an air of disappointment and scepticism by some commentators. Although many issues need to be addressed in its continuing development, gene therapy remains an exciting prospect for the future.
Gene therapy for cystic fibrosis
The tasks ahead include developing improved vectors and efficient delivery systems while avoiding adverse responses, discovering and targeting the correct cells, and achieving appropriate levels of expression in these cells over a lifetime. To this end the further development of animal models and continuing basic research into the cellular and molecular biology of this disease is crucial to achieve both enhanced gene delivery and expression and improved measurements of surrogate markers of clinical relevance.
Conclusions
Improvements over the last 25 years in the symptomatic treatment of cystic fibrosis lung disease have dramatically improved survival, but significant further improvements will require rational approaches based upon a more comprehensive understanding of the underlying disease pathogenesis. In this regard, the molecular cloning of the cystic fibrosis gene was a watershed and has precipitated dramatic changes in our perception of the disease. Nevertheless, it takes time and considerable basic research to translate gene discovery into new medicines and much remains to be understood about the intricacies of CFTR function and the sequelae of dysfunction. This new knowledge has, however, suggested a variety of radical and revolutionary approaches worthy of clinical investigation. The competition and collaboration between the various disciplines (microbiology, pharmacology and molecular biology) will ensure that these proposals and research findings are critically reviewed and refined, all to the benefit of the patient with cystic fibrosis and respiratory medicine in general.
References
Linked Articles
- Review series