Article Text

Abstract
Rationale Dendritic cells (DC) accumulate in the lungs of patients with idiopathic lung fibrosis, but their pathogenetic relevance is poorly defined.
Objectives To assess the role of the FMS-like tyrosine kinase-3 ligand (Flt3L)-lung dendritic cell axis in lung fibrosis.
Measurements and main results We demonstrate in a model of adenoviral gene transfer of active TGF-β1 that established lung fibrosis was accompanied by elevated serum Flt3L levels and subsequent accumulation of CD11bpos DC in the lungs of mice. Patients with idiopathic pulmonary fibrosis also demonstrated increased levels of Flt3L protein in serum and lung tissue and accumulation of lung DC in explant subpleural lung tissue specimen. Mice lacking Flt3L showed significantly reduced lung DC along with worsened lung fibrosis and reduced lung function relative to wild-type (WT) mice, which could be inhibited by administration of recombinant Flt3L. Moreover, therapeutic Flt3L increased numbers of CD11bpos DC and improved lung fibrosis in WT mice exposed to AdTGF-β1. In this line, RNA-sequencing analysis of CD11bpos DC revealed significantly enriched differentially expressed genes within extracellular matrix degrading enzyme and matrix metalloprotease gene clusters. In contrast, the CD103pos DC subset did not appear to be involved in pulmonary fibrogenesis.
Conclusions We show that Flt3L protein and numbers of lung DC are upregulated in mice and humans during pulmonary fibrogenesis, and increased mobilisation of lung CD11bpos DC limits the severity of lung fibrosis in mice. The current study helps to inform the development of DC-based immunotherapy as a novel intervention against lung fibrosis in humans.
- flt3l
- dendritic cells
- fibrosis
- lung
- MMP
Statistics from Altmetric.com
Key messages
What is the key question?
The role of the FMS-like tyrosine kinase-3 ligand (Flt3L)/DC axis in pulmonary fibrogenesis is unclear.
What is the bottom line?
We show that Flt3L protein is increased in human idiopathic pulmonary fibrosis lungs and mice with established lung fibrosis, and contributes to regulation of pulmonary fibrosis in mice through mobilisation of CD11bpos DC.
Why read on?
The current study helps to inform the development of Flt3L/DC-based immunotherapy as a novel intervention against pulmonary fibrosis in humans.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a diffuse parenchymal lung disease presenting with the histopathological feature of usual interstitial pneumonia, characterised by subepithelial fibroblastic foci, along with bibasilar subpleural honeycombing, as observed on high-resolution CT scans. The mean survival of IPF after diagnosis is 2–5 years, with a prevalence between 2 and 29 cases per 100 000 individuals in the general population, and more than 200 per 100 000 in the age group above 70.1 2
The pathogenesis of IPF is still poorly understood. In recent years, progress has been made regarding possible involvement of epithelial stem cell exhaustion, as well as repetitive injury of the alveolar epithelium triggering epithelial to mesenchymal transition pathways to contribute to IPF pathogenesis.3 4 At the same time, fibrotic lungs are infiltrated by immature DC which develop and proliferate from the bone marrow under regulation of the DC-specific growth factor FMS-like tyrosine kinase-3 ligand (Flt3L).5 Due to their principal localisation within the lung interstitial compartment, lung DC are strategically positioned to encounter collagen-rich interstitial extracellular matrix (ECM) deposition characterising the fibrogenic lung tissue remodelling process in IPF.6 However, previous reports also demonstrated that DC together with T cells and B cells may form ectopic lymphoid follicles in the lung of human IPF, where non-proliferating lymphocytes induce the maturation of DC, thus suggesting a role for DC and activated, non-proliferating lymphocytes contributing to chronic inflammation in interstitial lung fibrosis.7 8
In the current study, we aimed at elucidating the role of the Flt3L-lung DC axis in a mouse model of lung fibrosis triggered by adenoviral gene transfer of biologically active TGF-β1 (AdTGF-β1).9 By employing Flt3L-deficient mice (Flt3L KO) as well as diphteria toxin-treated (DT-treated) zDC+/DTR chimeric mice lacking major lung DC subsets,10 and CD103 KO mice lacking integrin αE (CD103) on the cell surface of CD103pos DC,11 as well as Batf3 KO mice lacking the CD103pos DC subset, we report that Flt3L contributes to limit the extent of lung fibrosis in response to AdTGF-β1 most likely through mobilisation and activation of CD11bpos DC but not CD103pos DC. Moreover, we show that therapeutic administration of Flt3L ameliorates lung fibrosis in this model.
Some of the results of these studies have been previously reported in the form of an abstract.12
Methods
All animal experiments were approved by local government authorities. The methods are outlined in detail in the online supplementary data.
Results
Recruitment and localisation of DC in lung tissue of wild-type mice with AdTGF-β1-induced lung fibrosis
We initially assessed collagen contents in lung tissue of wild-type (WT) mice exposed to empty control vector (Ad CL), or AdTGF-β1. Consistent with recent reports,13 we found significantly increased hydroxyproline levels in lungs of mice exposed to AdTGF-β1, relative to control vector exposed mice (online supplementary figure S1). Using flow cytometry, we next examined recruitment kinetics of the two major lung DC subsets, that is, CD11bpos DC and CD103pos DC in lungs of WT mice either left untreated, or exposed to control vector (Ad CL), or AdTGF-β1 (figure 1). As shown in Figure 1, particularly CD11bpos DC and, to a lesser extent, CD103pos DC accumulated in lungs of mice after AdTGF-β1 exposure, relative to control vector, with peak DC counts between days 14 and 28 (figure 1A–D). Notably, such significantly accentuated lung DC recruitment during establishing lung fibrosis was paralleled by increased serum levels of the DC-specific growth factor Flt3L, particularly in AdTGF-β1 but not control vector exposed mice (figure 1E). Examination of frozen lung tissue sections of mice with established lung fibrosis by immunofluorescence microscopy showed that lung DC (identified as low green autofluorescent, CD11cpos, MHCIIpos cells) localised in lung tissue of control vector exposed mice both in alveolar septae and the bronchovascular compartment (online supplementary figure S2). In the lungs of AdTGF-β1-exposed mice, increased lung DC accumulation was observed within thickened alveolar septae of fibrotic lung lesions (figure 1F, yellow arrows in quadrant 4).
Supplemental material

Recruitment kinetics and localisation of CD11bpos DC and CD103pos DC subsets in lungs of WT mice exposed to control vector or AdTGF-β1. (A and B) Immunophenotypic analysis of CD11bpos and CD103pos DC in lungs of (A) control vector (AD Cl) or (B) AdTGF-β1-exposed mice (day 14), as detailed in methods. (C and D) Quantification of lung (C) CD11bpos DC and (D) CD103pos DC in the lungs of untreated control mice (Cl, grey dots), or control vector exposed mice (white dots) or AdTGF-β1-exposed mice (black dots), according to FACS-based gating of DC subsets as outlined in (A and B). (E) Murine Flt3L protein levels in serum of untreated WT mice, or WT mice exposed to control vector (white dots) or AdTGF-β1 (black dots) at the indicated time points. Data are shown as median ± IQR of n=5–10 (C and D), or n=5–8. (E) mice per time point and treatment group and are representative of two independent experiments. *p<0.05, **p<0.01, ***p<0.001 versus control vector using Mann-Whitney U test. (F) Immunofluorescence analysis of lung DC in frozen lung tissue sections of WT mice exposed to AdTGF-β1 for 14 days. Orange arrows indicate low green autofluorescent, CD11c-AF647pos, MHCII-AF568pos lung DC within fibrotic lung lesions. A representative image out of four independent analyses is shown. Original magnification, ×20. FACS, fluorescence-activated cell sorting; Flt3L, FMS-like tyrosine kinase-3 ligand; WT, wild type.
Next, we determined levels of Flt3L protein and lung DC distribution in patients with progressive fibrotic interstitial lung disease (ILD). As shown in figure 2A, significantly increased levels of Flt3L protein were detected in serum as well as subpleural lesions of explanted lung tissue specimen from patients with end-stage lung fibrosis, relative to disease controls (figure 2A,B; table 1). Moreover, using immunofluorescence analysis of frozen lung tissue sections, CD11cpos/MHCIIpos lung DC were detected in fibrotic lung lesions from patients with ILD compared with disease controls, where lung DC were observed in alveolar septae of the lung (figure 2C, online supplementary figure S3).

Analysis of Flt3L protein levels in serum and lung tissue of patients with interstitial lung fibrosis. (A and B) Quantification of human Flt3L protein in serum from patients with ILD or disease controls (CL) (A), or in explant lung tissue from patients with ILD or CL, as indicated (B). Data are shown as median ±IQR of n=10–31 (A), or n=10–18 (B) patients with ILD or disease controls, as indicated. *p<0.05, **p<0.01 versus disease control using Mann-Whitney U test. (C) Representative immunofluorescence analysis of CD11c-AF647pos/MHCII-AF568pos dendritic cells in explant lung tissue from patients with ILD. Orange arrows in quadrant (Q) 4 display overlays of immunofluorescence signals of lung DC taken from quadrants 2 (DAPI+CD11c-AF647) and 3 (DAPI+MHCII-AF568), as indicated. Original magnification ×20. Flt3L, FMS-like tyrosine kinase-3 ligand; ILD, interstitial lung disease
Patient characteristics
Effect of Flt3L deficiency on AdTGF-β1-induced lung fibrosis
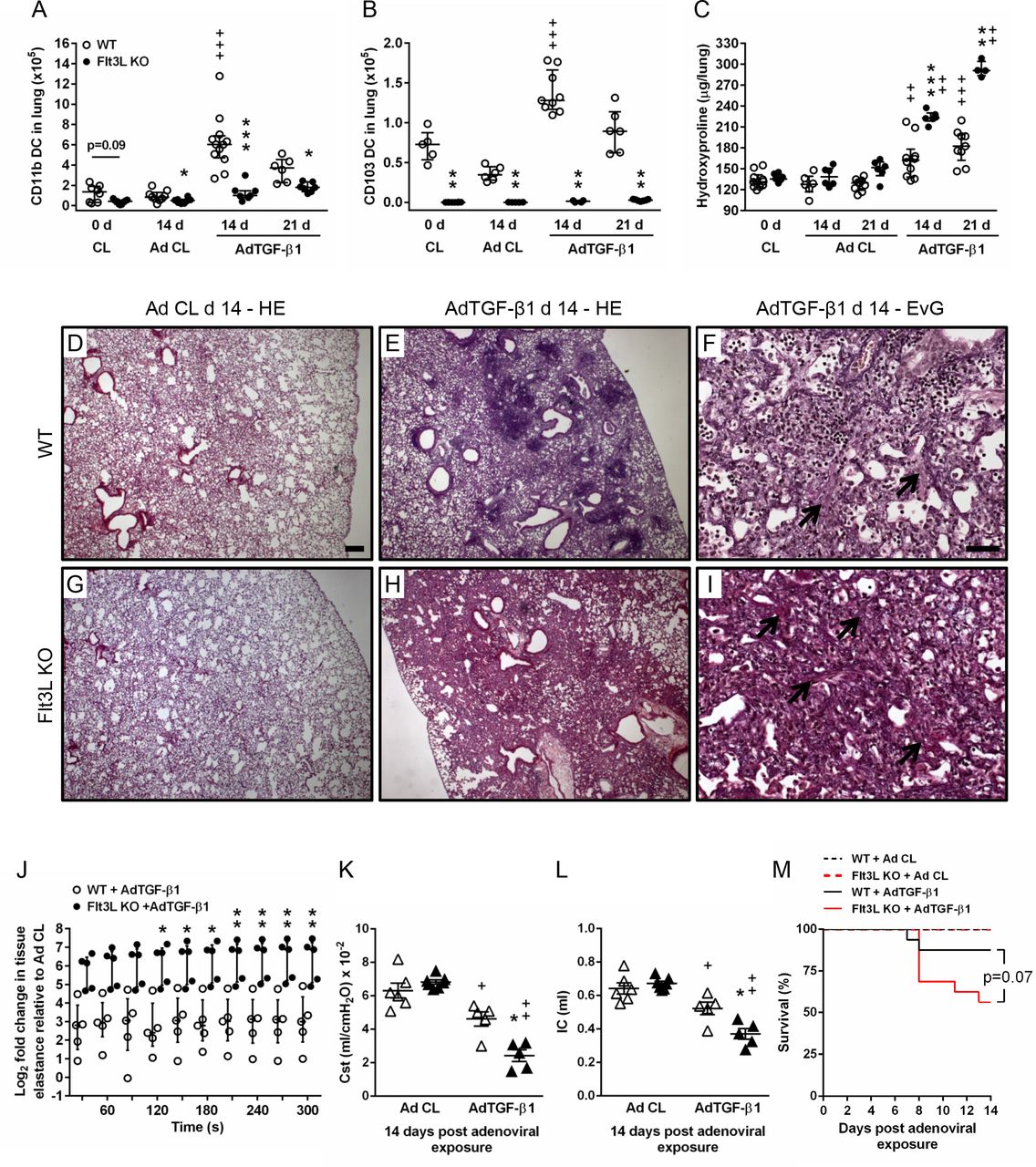
We next examined the effect of Flt3L deletion on lung DC subset accumulation and fibrosis in mice exposed to AdTGF-β1. As shown in figure 3A,B and online supplementary figure S4, lung DC subset counts were significantly reduced in Flt3L KO mice under baseline conditions. Moreover, numbers of CD11bpos DC and CD103pos DC were significantly diminished in AdTGF-β1-exposed Flt3L KO mice, compared with AdTGF-β1-exposed WT mice (figure 3A,B, Figure S4C,D). Importantly, Flt3L KO mice responded with significantly increased lung collagen contents at days 14 and 21, relative to WT mice exposed to AdTGF-β1 (figure 3C). Histopathological examination of lung tissue sections demonstrated thickened alveolar septae with interstitial fibrosis evidenced by increased collagen deposition together with inflammatory lymphoplasmacellular infiltrates in AdTGF-β1-exposed WT mice (figure 3D–F). This phenotype was substantially aggravated in AdTGF-β1-exposed Flt3L KO mice, which exhibited hilar accentuated interstitial lung fibrosis with accompanying elastosis and hyperplastic type II alveolar epithelial cells (figure 3G–I).

Effect of Flt3L deficiency on AdTGF-β1-induced lung fibrosis in mice. (A–C) Quantification of lung CD11bpos DC (A) and CD103pos DC (B), and collagen contents (C) in lungs of WT mice and Flt3L KO either left untreated (0 D), or exposed to either control vector (white dots) or AdTGF-β1 (black dots) at the given time points. (D–I) Histopathology of lung tissue sections from WT mice and Flt3L KO mice exposed to AdCL (D and G) or AdTGF-β1 (E, F, H and I) (day 14) stained with either haematoxylin–eosin (D, E, G and H), or trichrome (Elastica-van-Gieson) (F and I), as indicated. Black arrows indicate collagen fibre deposition. Representative images of 3–4 mice per experimental group are shown at a magnification of ×2.5 (D, E, G and H) and ×20 (F,I). Scale bar, 200 µm (D, E, G and H), and 50 µm (F,I). (J–L) Quantification of tissue elastance [H] (J) relative to control vector, or static compliance (K), or IC (L) in WT mice or Flt3L KO mice at day 14 post-adenoviral exposure. (M) Survival of WT versus Flt3L KO mice after AdCL or AdTGF- β1 exposure. Data are shown as median ±IQR of n=6–12 (A), n=4–9 (B), n=4–10 (C), n=5 (J), n=5–7 (K and L) or n=10–15 (M) mice per time point and treatment group and are representative of three (A–C, M) or two independent experiments (J–L). *p<0.05, **p<0.01, ***p<0.001 versus WT under the same treatment conditions;+p<0.05, ++p<0.01 and +++p<0.001 versus respective control vector treatment (Mann-Whitney U test [A–C,K and L], two-way analysis of variance [J], log-rank test [M]). Flt3L, FMS-like tyrosine kinase-3 ligand; IC, inspiratory capacity; WT, wild type.
Flt3L KO mice exposed to AdTGF-β1 demonstrated similar numbers of lung macrophages, B cells and T cells compared with WT mice (online supplementary figure S5A-C), but numbers of natural killer (NK) cells were significantly diminished in AdTGF-β1-exposed Flt3L KO mice (online supplementary Figure S5J). However, depletion of NK cells with antibody NK1.114 (online supplementary figure S5D-I) did not alter the degree of lung fibrosis on day 14 post-treatment as observed in isotype antibody treated, AdTGF-β1-exposed mice (online supplementary figure S5K), thus ruling out a major role for NK cells in the currently employed AdTGF-β1-induced lung fibrosis model.
Assessment of respiratory mechanics in WT and Flt3L KO mice exposed to control vector or AdTGF-β1 for 14 days showed significantly increased lung tissue elastance in Flt3L KO mice as compared with WT mice exposed to AdTGF-β1 at steady state (210–300 s) (figure 3J). Accordingly, static compliance (Cst) and inspiratory capacity (IC) were significantly reduced in Flt3L KO mice compared with WT mice after AdTGF-β1 exposure, confirming that lung function was severely impaired in Flt3L KO mice relative to WT mice exposed to AdTGF-β1 (figure 3K,L). Consistent with this finding, Flt3L KO mice exhibited strongly increased mortality in response to AdTGF-β1, relative to AdTGF-β1-exposed WT mice (p=0.07) (figure 3M). Together, these data illustrate that the Flt3L-lung DC axis contributes to regulation of lung collagen deposition in AdTGF-β1-exposed mice, and lack of Flt3L aggravates lung fibrosis.
Effect of Flt3L administration on AdTGF-β1-induced lung fibrosis in Flt3L KO mice
We then examined whether the observed phenotype of exacerbated lung fibrosis in Flt3L KO mice would be improved by treatment of Flt3L KO mice with recombinant Flt3L (rFlt3L). As shown in figure 4, Flt3L KO mice responded with substantially expanded CD11bpos DC and CD103pos DC subsets on rFlt3L application, compared with vehicle-treated Flt3L KO mice (figure 4A–C). At the same time, pre-treatment with rFlt3L resulted in significantly reduced lung collagen contents in Flt3L KO mice exposed to AdTGF-β1 compared with vehicle-pre-treated Flt3L KO mice (figure 4D). Moreover, we observed significantly reduced lung tissue elastance in Flt3L KO mice pre-treated with rFlt3L relative to vehicle-treated Flt3L KO mice exposed to AdTGF-β1 (figure 4E). Correspondingly, Cst and IC were significantly increased in rFlt3L-treated Flt3L KO mice compared with vehicle-treated Flt3L KO mice at day 14 post AdTGF-β1 exposure (figure 4F,G). In addition, pre-treatment of Flt3L KO mice with rFlt3L led to substantially (~40 %) reduced mortality, relative to vehicle pre-treated Flt3L KO mice exposed to AdTGF-β1 for 14 days (figure 4H), although this difference did not reach statistical significance (p=0.07). These data illustrate that rFlt3L application significantly improves lung fibrosis (and mortality) in Flt3L KO mice exposed to AdTGF-β1.
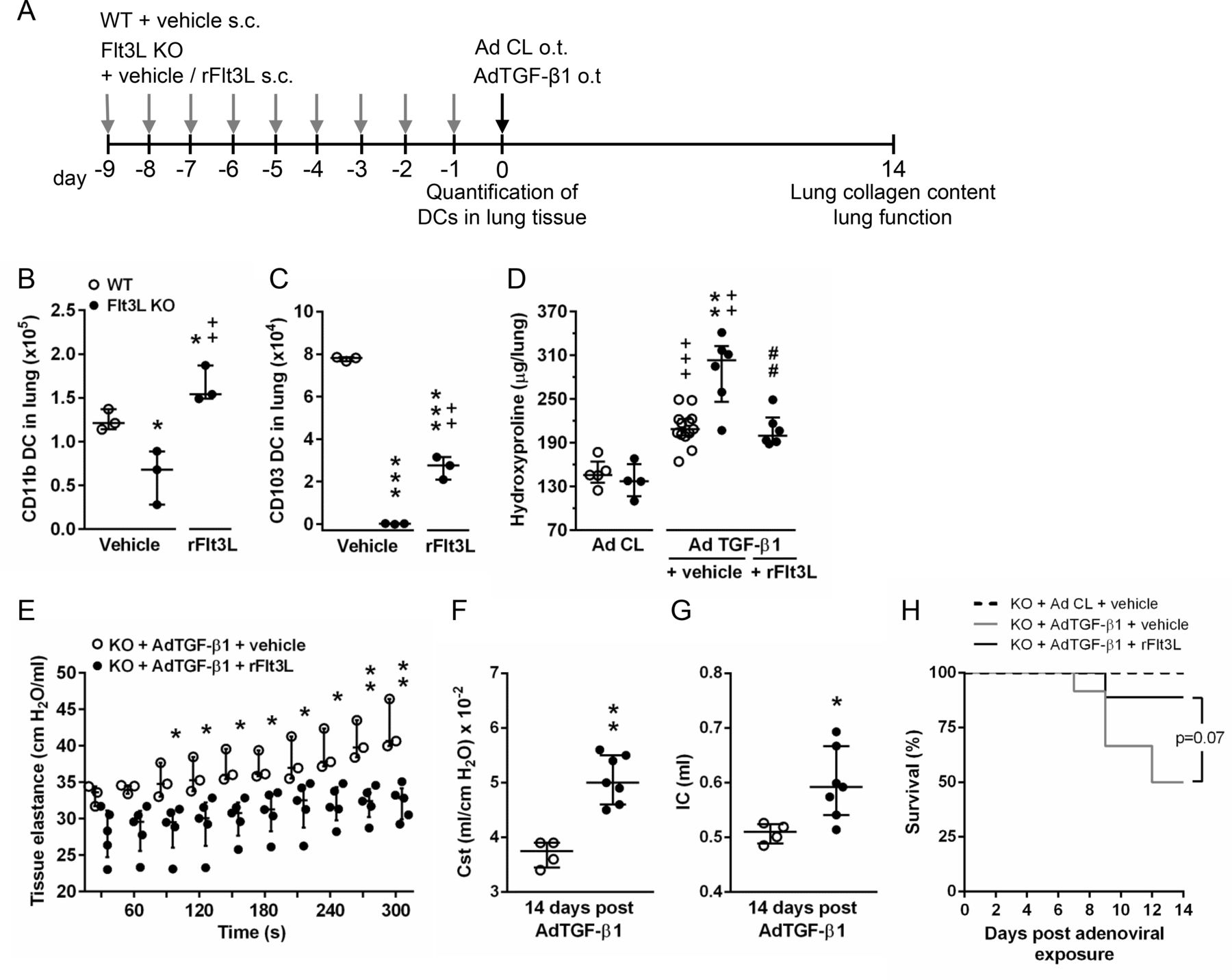
Effect of recombinant Flt3L treatment on lung fibrosis in Flt3L KO mice exposed to AdTGF-β1. (A) Experimental profile. WT or Flt3L KO mice were treated daily with vehicle or rFlt3L for nine consecutive days. Subsequently, mice were subjected to lung DC quantification, or mice were exposed to control vector (AD Cl) or AdTGF-β1 (day 0) for determination of lung collagen deposition or lung function at day 14. (B,C) Quantification of CD11bpos DC (B) and CD103pos DC (C) of WT or Flt3L KO mice at 9 days post vehicle or rFlt3L treatment. (D) Hydroxyproline levels in lung tissue of vehicle-treated or rFlt3L-treated WT and Flt3L KO mice at day 14 post-adenoviral exposure. (E–G) Quantification of tissue elastance [H] (E), static compliance (F) and inspiratory capacity (G) in Flt3L KO mice at day 14 post-adenoviral exposure after vehicle or rFlt3L treatment. (H) survival of Flt3L KO mice pre-treated with vehicle or rFlt3L and exposed to AD Cl or AdTGF- β1. Data are shown as median ±IQR of n=3 (B,C), n=4–13 (D), n=3–5 (E), n=4–7 (F,G), or n=7–12 (H) per time point and treatment group and are representative of two independent experiments. *p<0.05 and ***p<0.001 versus WT+vehicle,++p<0.01 versus Flt3L KO +vehicle (B and C).++p<0.01 and +++p<0.001 versus respective control vector treatment, **p<0.01 versus WT+AdTGF-β1+vehicle, and ##p<0.01 versus Flt3L KO +AdTGF-β1+vehicle (D). *p<0.05 and **p<0.01 versus Flt3LKO+AdTGF-β1+vehicle (E–G). (Mann-Whitney U test [B–D, F and G], two-way analysis of variance [E], log-rank test [H]). Flt3L, FMS-like tyrosine kinase-3 ligand; rFlt3L, recombinant Flt3L; WT, wild type.
Treatment with rFlt3L improves lung fibrosis in WT mice exposed to AdTGF-β1
Having demonstrated that treatment with rFlt3L significantly attenuated lung tissue remodelling and mortality in AdTGF-β1-exposed Flt3L KO mice, we next questioned whether therapeutic Flt3L administration would also impact on lung fibrosis in WT mice exposed to AdTGF-β1. WT mice responded to therapeutic application of Flt3L (initiated on day 4 until day 13 post-AdTGF-β1) with a significantly expanded CD11bpos and CD103pos DC subset accumulation in lung tissue, illustrating that increased bioavailability of Flt3L triggered DC accumulations in lung tissue of WT mice with established lung fibrosis (figure 5A–C). Importantly, therapeutic treatment with rFlt3L significantly reduced lung collagen contents in WT mice exposed to AdTGF-β1 (figure 5D). These data support the view that Flt3L therapy is efficacious to attenuate established lung fibrosis in WT mice.

Effect of rFlt3L treatment on lung fibrosis in WT mice exposed to AdTGF-β1. (A) Experimental profile. WT mice were exposed to control vector (AD Cl, grey dots), or AdTGF-β1 and were treated daily with vehicle or rFlt3L from day 4 until day 13 after adenoviral exposure, as indicated (B and C). Quantification of CD11bpos DC (B) and CD103pos DC (C) of vehicle or rFlt3L-treated WT mice exposed to AdTGF-β1 for 14 days. (D) Hydroxyproline levels in lung tissue of vehicle- or rFlt3L-treated WT mice at day 14 post-adenoviral exposure, as indicated. Data are shown as median ±IQR of n=3 (B,C) or n=3–6 (D) mice and are representative of two independent experiments. *p<0.05, **p<0.01, ***p<0.001 versus AdTGF-β1+vehicle,+p<0.05 versus Ad Cl +vehicle using Mann-Whitney U test. rFLT3L: Flt3L, FMS-like tyrosine kinase-3 ligand; recombinant Flt3L; WT, wild type.
Effect of DC depletion on AdTGF-β1-induced lung fibrosis in WT and zDC+/DTR chimeric mice
We next examined the effect of lung DC depletion on AdTGF-β1-induced lung fibrosis in zDC+/DTR chimeric mice allowing diphtheria toxin-induced specific DC depletion. As shown in figure 6, DT treatment of zDC+/DTR chimeric mice led to a significant depletion of lung CD11b DC and CD103 DC, relative to WT chimeric mice treated with DT (figure 6A–D). Importantly, DT-induced specific DC depletion in zDC+/DTR chimeric mice exposed to AdTGF-β1 also led to progressive lung fibrosis, relative to DT-treated, AdTGF-β1-exposed WT chimeric mice (figure 6E). At the same time, DT treated, AdTGF-β1-exposed zDC+/DTR chimeric mice exhibited significantly increased lung tissue elastance and reduced Cst along with a significantly reduced IC compared with DT-treated chimeric WT mice, overall demonstrating significantly impaired lung function in response to DC depletion in AdTGF-β1-exposed chimeric zDC+/DTR mice, relative to chimeric WT mice (figure 6F–H). Collectively, these data provide additional support for a contribution of lung DC in the regulation of pulmonary fibrosis.
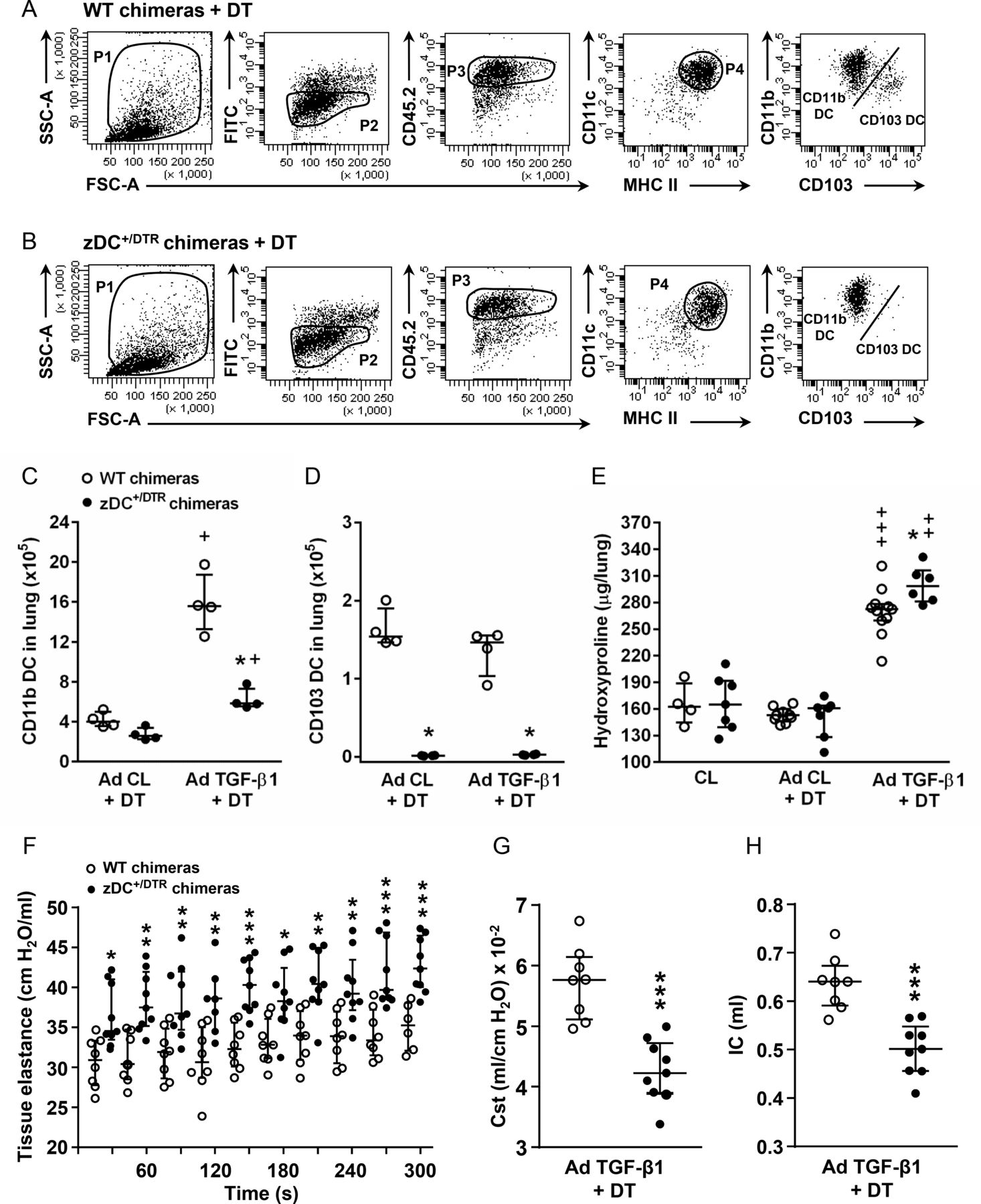
Effect of DC depletion on AdTGF-β1-induced lung fibrosis in WT chimeric and zDC+/DTR chimeric mice. (A and B) Gating of lung donor-type CD45.2pos CD11bpos DC and CD103pos DC subsets in lungs of (A) WT chimeric mice and (B) zDC+/DTR chimeric mice treated with control vector (AD Cl)+DT or AdTGF-β1+DT for 14 days. (C and D) Quantification of donor-type (C) CD45.2pos CD11bpos DC and (D) CD45.2pos CD103pos DC at day 14 post-treatment. (E) Collagen contents in lung tissue of untreated chimeric mice (CL), or respective chimeric mice treated with control vector (AD Cl)+DT or AdTGF-β1+DT at day 14 post-adenoviral exposure. (F–H) Assessment of lung mechanics (tissue elastance H in (F), and static compliance in (G) and inspiratory capacity in (H)) in AdTGF-β1 exposed and DT-treated WT or zDC+/DTR chimeric mice at day 14 post-treatment. Data are shown as median ±IQR of n=4 (C and D), or n=4–11 (E), or n=7–9 mice (F–H) and are representative of two to three independent experiments. *p<0.05, **p<0.01, ***p<0.001 versus respective treatment in WT chimeric mice;+p<0.05,++p<0.01,+++p<0.001 versus respective AD Cl +DT treatment using Mann-Whitney U test. Cst, static compliance; WT, wild type.
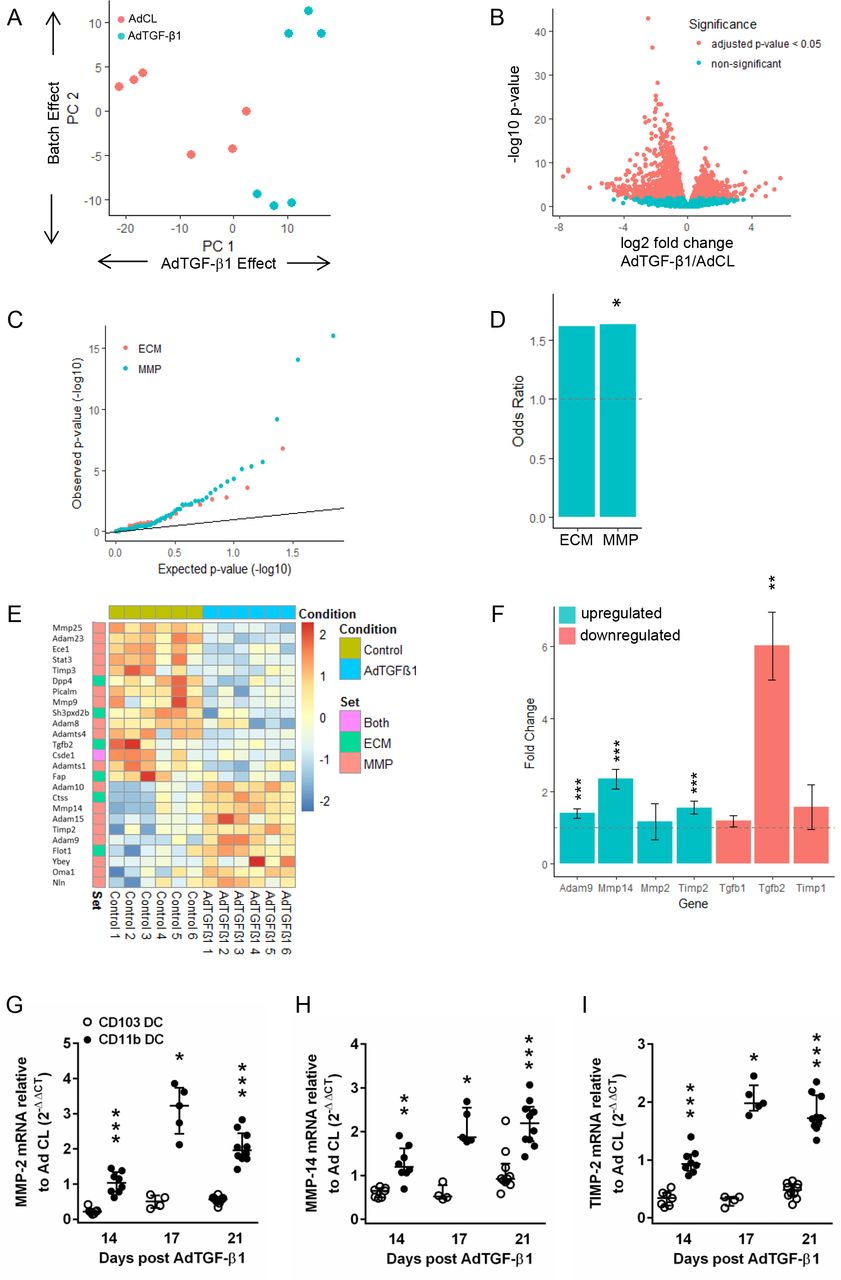
RNA-sequencing analysis of CD11bpos DC from mice with established lung fibrosis reveals significantly enriched differentially expressed genes in ECM and matrix metalloprotease gene categories
We next performed next generation sequencing (NGS)-based transcriptomic profiling of the major CD11bpos DC subset to characterise its potential antifibrogenic profile. As shown in figure 7A, principal component analysis revealed that the majority of signal variation observed between groups was due to treatment conditions (Ad CL vs AdTGF-β1). A total of 2279 differentially expressed genes (DEGs) were identified using an adjusted p value threshold of 0.05. Of these, the majority (1418) were actually downregulated in CD11bpos DC of AdTGF-β1 versus Ad CL-exposed mice (figure 7B). Plotting the observed p values for ECM and matrix metalloprotease (MMP) genes against the expected p values under the null hypothesis (figure 7C) showed enrichment for low p values in ECM and MMP genes. Both gene sets exhibited similar levels of over-representation for DEGs (OR of 1.6, figure 7D) but this was only statistically significant (p value<0.05) for MMP genes, due to the lower number of genes in the ECM set. AdTGF-β1 treatment caused downregulation of some ECM and MMP markers, whereas others were significantly upregulated in CD11bpos DC (figure 7E). More specifically, a disintegrin-like metalloprotease-9 (ADAM9) as well as MMP2, its activator MMP14 and its inhibitor TIMP-2 collectively serving as a membrane-anchored multimeric proteolytic complex15 were among those genes upregulated, while profibrotic TGF-β1 and TGF-β2 as well as TIMP-1 were downregulated in response to AdTGF-β1 (figure 7F). In line with this, real-time PCR analysis of MMP-2, MMP14 and TIMP-2 confirmed its significant upregulation in sorted CD11bpos DC but not CD103pos DC of AdTGF-β1-exposed mice (figure 7G–I; online supplementary figure S6), thus supporting a role for CD11bpos DC rather than CD103pos DC in pulmonary fibrogenesis.
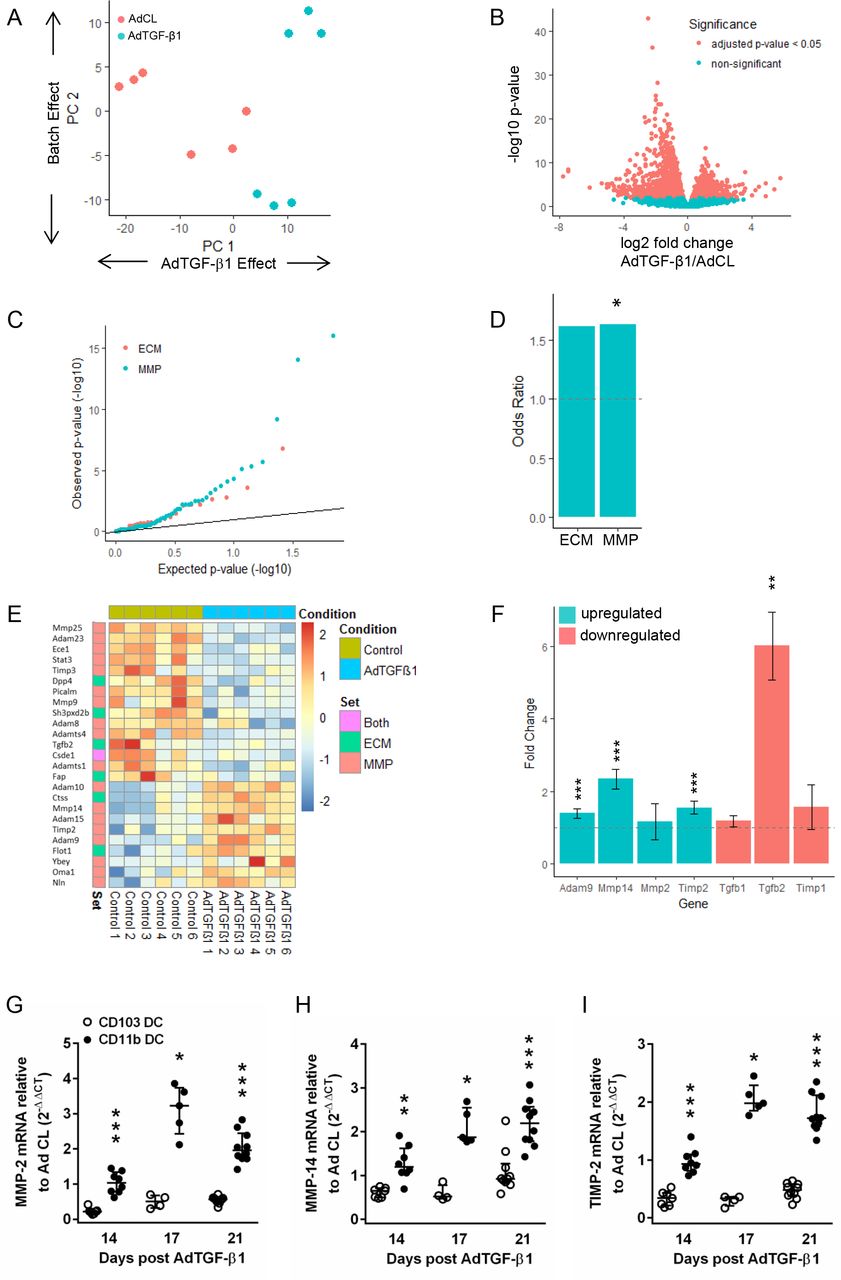
Differential gene expression analysis of CD11bpos dendritic cells from AdTGF-β1 and control vector treated mice. CD11bpos DC were sorted from lungs of mice exposed to AdTGF-β1 or control vector (day 14 post-treatment), followed by RNA-sequencing analysis, as outlined in methods. (A) Principle component analysis indicating that the majority of signal variation is associated with the treatment conditions. (B) Volcano plot indicating the relationship between significance and fold change for 2279 differentially regulated genes (C–E) depicting the significance of changes in ECM and MMP gene expression. (C) Q–Q plot of expected versus observed p values indicating enrichment for low p values in ECM and MMP genes. (D) OR for ECM and MMP genes with statistical significance observed for the MMP gene cluster. (E) Heat map of upregulated and downregulated ECM and MMP genes in CD11b DC from AdTGF-β1 versus control vector exposed mice (day 14 post-treatment). (F) Fold up-/downregulation of ADAM9, MMP-2, MMP-14, TIMP1, TIMP2, TGF-β1 and TGF-β2 genes in CD11bpos DC of AdTGF-β1 relative to control vector exposed mice (day 14 post-treatment), as indicated. (G–I) Real-time PCR analysis of MMP2 (G), MMP14 (H) and TIMP-2 (I) gene expression in CD11bpos DC and CD103pos DC of AdTGF-β1 relative to control vector exposed mice (fold change) on day 14 post-treatment. Data are shown as median ±IQR of n=4–10 mice (G–I) and are representative of two independent experiments. *p<0.05, **p<0.01, ***p<0.001 relative to control vector treatment (D and F). *p<0.05, **p<0.01, ***p<0.001 relative to CD103pos DC (G–I). (Mann-Whitney U test). ECM: extracellular matrix; MMP, matrix metalloprotease.
Effect of CD103 deficiency or CD103 DC deficiency on lung fibrosis in mice exposed to AdTGF-β1
To further characterise the role of CD103pos DC in lung fibrosis in the described model system, CD103 KO mice or Batf3 KO mice lacking the CD103pos DC subset were exposed to AdTGF-β1. As shown in figure 8A,B, flow cytometry revealed that integrin αE (CD103) was not expressed on the respective DC subset in the lungs of CD103 KO mice compared with WT mice, while the respective (CD103neg) myeloid cell population was still detectable in CD103 KO mice (figure 8A,B, upper and lower dot plots). In contrast, Batf3 KO mice were found to lack the CD103pos DC subset (figure 8A vs 8C). Flow cytometric quantification of DC subsets in lungs of WT and CD103 KO mice or Batf3 KO mice showed similar levels of CD11bpos DC but lack of CD103pos DC in both knockout mouse strains opposed to WT mice (figure 8D,F).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of CD103 deficiency or CD103 DC deficiency on AdTGF-β1-induced lung fibrosis in mice. (A–C) Flow cytometric gating strategy for immunophenotypic identification of CD11bpos, CD103neg DC (CD11bpos dC) and CD11bneg, CD103pos DC (CD103pos dC) subsets in lungs of untreated WT mice (A), or CD103 KO mice (B), or Batf3 KO mice (C). (D) Quantification of DC subsets in lungs of WT mice and CD103 KO mice (day 14 post-AdTGF-β1). (E) Determination of collagen contents in lung tissue of untreated, or control vector, or AdTGF-β1-exposed WT versus CD103 KO mice, as indicated. (F) Quantification of DC subsets in lungs of WT mice and Batf3 KO mice (day 14 post- AdTGF-β1). (G) Collagen contents in lung tissue of untreated (CL), or control vector (AD Cl), or AdTGF-β1-exposed WT versus CD103 DC-deficient (Batf3 KO) mice. Data are shown as median ±IQR of n=3 (D,F), n=4–6 (E), or n=3–6 (G) mice, and are representative of at least two independent experiments. *p<0.05 versus respective WT; and +p<0.05, ++p<0.01 versus respective control vector using Mann-Whitney U test. WT, wild type
We then examined the impact of CD103 deficiency or CD103pos DC deficiency on AdTGF-β1-induced lung fibrosis. As shown in figure 8E and G, CD103 KO mice (figure 8E) and Batf3 KO mice (figure 8G) presented similar collagen contents in their lungs after AdTGF-β1 exposure as WT mice, with no significant differences between groups. This finding confirmed that neither integrin αE (CD103) nor CD103 DC do contribute to lung fibrogenesis in mice exposed to AdTGF-β1.
Discussion
The pathogenetic impact of DC recruitment in interstitial lung fibrosis has not been characterised yet. Using a well-defined murine model of lung fibrosis induced by adenoviral exposure to biologically active TGF-β1, we identified an important role for the Flt3L-lung DC axis in limiting the degree of lung fibrosis in mice, which is supported by the following findings: (1) Increased levels of Flt3L protein are consistently found in both mice with established lung fibrosis and patients with lung fibrosis, which is accompanied by increased recruitment of lung DC to the sites of fibrotic remodelling. (2) Flt3L KO mice or DT-treated transgenic zDC+/DTR mice lacking lung DC demonstrate aggravated lung fibrosis, which can be reversed by treatment with recombinant human Flt3L. (3) Therapeutic application of human Flt3L also improves the degree of AdTGF-β1-induced lung fibrosis in WT mice. (4) NGS analysis of CD11bpos DC collected from fibrotic mouse lungs reveals an antifibrotic transcriptomic signature of this DC subset, collectively supporting the view that the Flt3L-lung DC axis is involved in limiting pulmonary fibrogenesis. DC-based immunotherapy is a novel approach in oncology, and various clinical trials have explored the safety and efficacy of recombinant human Flt3L in healthy volunteers and in cancer therapy to safely trigger DC expansion in humans.16–19 This, together with our mechanistic data, may open a novel therapeutic avenue for the management of IPF.
Flt3L is the main growth factor driving lymphoid and non-lymphoid classical DC differentiation from committed bone marrow progenitors called pre-DC.20 21 Once Flt3-expressing pre-DCs have arrived in peripheral tissues, they differentiate into the various subsets of classical dendritic cells (cDCs) in the presence of Flt3L.20 We observed increased serum levels of Flt3L in AdTGF-β1-exposed WT mice, and increased levels of human Flt3L in serum and subpleural tissue of explanted lungs from patients with interstitial lung fibrosis (mostly IPF), which was accompanied by accumulation of lung DC in fibrotic lungs while lung tissue specimen collected from central compartments of the same lungs did not exhibit elevated levels of Flt3L protein. This differential release of Flt3L may be of interest, since the primary focus of fibrotic remodelling in IPF lungs is in the peripheral, subpleural rather than central areas of the lung, where Flt3L-induced DC mobilisation, according to the current study, would serve best to limit fibrotic remodelling.
Regarding the question which cell types contribute to Flt3L release in normal and fibrotic lungs, several studies demonstrated that Flt3L as a haematopoietic growth factor is expressed in virtually every organ system examined so far based on mRNA, including the lung,22 23 with low baseline levels in normal human blood serum.24 Particularly, lymphocytes and haematopoietic stromal cells are the major producers of Flt3L.25 26 Moreover, many other cell types (and cell lines) have been reported to express Flt3L, including endothelial cells27 and tumour-derived NK cells.28 Based on the fact that most of these cell types are found in normal and fibrotic lungs, it is currently impossible to say to what extent which cell type contributes to Flt3L observed in fibrotic lungs. Regarding elevated Flt3L levels observed in the lungs of patients with IPF and mice with established fibrosis, the most likely explanation is its systemic upregulation, which reaches the lungs via the blood stream. In addition, lymphoplasmacellular infiltrates observed in the lungs of patients with IPF and mice, according to the cellular sources of Flt3L outlined above, will further contribute to increased Flt3L levels in lung parenchyma of patients with ILD.
The presented data strongly argue for a contribution of the Flt3L-lung DC axis in limiting the degree of ECM deposition during lung fibrosis, not least in view of the fact that lung DC are strategically located in the lung interstitium with direct access to ECM within the parenchyma. In line with this, RNA-sequencing analysis of CD11bpos DC revealed significant upregulation of markers of ECM turnover such as a ADAM9, which is known to cleave ECM components fibronectin, laminin and elastin,29 30 as well as MMP-2 and MMP-14 and its tissue inhibitor TIMP-2. Notably, MMP14, also designated as MT1-MMP, has been shown to cleave native type I and III collagens typical of specific collagenases.31 Due to the very limited amount of sorted CD11bpos DC, it was not possible to additionally examine the respective proteomic signature of the CD11bpos DC subset in the current model system. Notably, although MMP2 expression is observed in IPF lungs, examination of the role of MMP2 in lung fibrosis has not been reported thus far.31 32 This is most likely because deficiency of MMP2, designated as gelatinase A, might simply be compensated by MMP9, also designated as gelatinase B, thus calling for respective experiments in MMP2/9 double KO mice, which was beyond the scope of the current study.
Previous reports showed a role for DC to regulate carbon tetrachloride or thioacetamide/leptin-induced hepatic fibrosis in transgenic CD11c+/DTR mice, where conditional DC depletion achieved by DT application delayed liver fibrosis regression.33 34 Other reports in mouse models of bleomycin-induced lung fibrosis demonstrated that pharmacological inactivation of DC with the immunomodulatory VAG539 inhibiting CD86 and MHC II expression on DC and B cells resulted in reduced fibrosis.35 In the current model system, we found that CD103pos DC of mice exposed to AdTGF-β1 did not present differential expression of MMP activation markers during established lung fibrosis. Consistent with these data, both CD103 KO mice and Batf3 KO mice lacking the CD103pos DC subset responded with similar fibrosis to AdTGF-β1 exposure as WT mice. Another study demonstrated increased bleomycin-induced fibrosis in CD103 KO mice compared with WT mice while suggesting that interleukin 10 released from CD103pos DC may limit the degree of bleomycin-induced fibrosis.11 Differences between the employed mouse models of lung fibrosis may at least partially explain why we did not find a role for CD103 to regulate pulmonary fibrogenesis neither in CD103 KO nor Batf3 KO mice.
In summary, the data of the current study show that Flt3L is elevated in mice with established lung fibrosis as well as in patients with lung fibrosis and contributes most likely through lung DC mobilisation to limit the degree of lung fibrosis. These findings underline the significance of specific growth factors of the myeloid compartment for the regulation of pulmonary fibrogenesis. The fact that this pathway is already explored in oncology for DC-based immunotherapy, and the availability of several clinical grade compounds, suggests that this might be a promising new avenue for future therapeutic interventions in IPF.
Acknowledgments
Sequencing data (RNA-Seq) were generated by the Research Core Unit Genomics at Hannover Medical School.
References
Footnotes
MRK and UAM are joint senior authors.
Contributors MTT carried out experiments, analysed the data and wrote the manuscript; FA, RM, JS, LS, LK, ELR and MK carried out experiments, DJ and DSD analysed the data, AP, TW, JG, MK and UAM designed the study, analysed the data and wrote the manuscript.
Funding The current study was financially supported by the Federal Ministry of Education and Research in support of the German Center for Lung Research, partner site Breath (Biomedical Research in Endstage and Obstructive Lung Disease Hannover).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.