Article Text

Abstract
Background Prolonged controlled mechanical ventilation (CMV) in humans and experimental animals results in diaphragm fibre atrophy and injury. In animals, prolonged CMV also triggers significant declines in diaphragm myofibril contractility. In humans, the impact of prolonged CMV on myofibril contractility remains unknown. The objective of this study was to evaluate the effects of prolonged CMV on active and passive human diaphragm myofibrillar force generation and myofilament protein levels.
Methods and results Diaphragm biopsies were obtained from 13 subjects undergoing cardiac surgery (control group) and 12 brain-dead organ donors (CMV group). Subjects in each group had been mechanically ventilated for 2–4 and 12–74 h, respectively. Specific force generation of diaphragm myofibrils was measured with atomic force cantilevers. Rates of force development (Kact), force redevelopment after a shortening protocol (Ktr) and relaxation (Krel) in fully activated myofibrils (pCa2+=4.5) were calculated to assess myosin cross-bridge kinetics. Myofilament protein levels were measured with immunoblotting and specific antibodies. Prolonged CMV significantly decreased active and passive diaphragm myofibrillar force generation, Kact, Ktr and Krel. Myosin heavy chain (slow), troponin-C, troponin-I, troponin-T, tropomyosin and titin protein levels significantly decreased in response to prolonged CMV, but no effects on α-actin, α-actinin or nebulin levels were observed.
Conclusions Prolonged CMV in humans triggers significant decreases in active and passive diaphragm myofibrillar force generation. This response is mediated, in part, by impaired myosin cross-bridge kinetics and decreased myofibrillar protein levels.
- Respiratory Muscles
- Assisted Ventilation
Statistics from Altmetric.com
Key messages
What is the key question?
Does prolonged controlled mechanical ventilation (CMV) in humans alter the contractile performance and myofilament protein expression in diaphragm myofibrils?
What is the bottom line?
Prolonged CMV in humans significantly decreases active and passive diaphragm myofibrillar force generation and significantly attenuates myofilament protein levels including those of myosin heavy chain, myosin light chain, troponin-C, I and T as well as titin.
Why read on?
This study implies that prolonged CMV in humans elicits significant impairment of active and passive performance by diaphragm myofibrils as a result of increased degradation of essential myofilament proteins by the proteasome and autophagy proteolytic pathways.
Introduction
Mechanical ventilation (MV) is a two-edged sword. On one hand, it is a life-saving procedure that is used in intensive care units to manage patients with respiratory failure and other pathologies such as cardiac failure and stroke. On the other hand, MV is associated with numerous complications, including ventilator-induced lung injury.1 In the past several years, many studies have indicated that use of the controlled mode of MV (CMV), where spontaneous diaphragm activity is inhibited for relatively long periods of time, results in the development of a condition known as ventilator-induced diaphragm dysfunction (VIDD).2 VIDD is associated with diaphragm muscle fibre atrophy, ultrastructure injury and depressed muscle force-generating capacity. Clinically, VIDD is a distinct entity from other causes of diaphragm dysfunction such as critical illness myopathy, reduced O2 delivery, electrolyte imbalance and steroid administration. For patients with acute respiratory failure, successful weaning from MV is largely determined by the work of breathing (dependent on lung mechanics) and the ability of the inspiratory muscles in general, and the diaphragm in particular, to cope with the work of breathing.3–5 Therefore, the development of VIDD is likely to contribute to weaning failure and results in prolonged durations of stay in intensive care units.
Animal studies have revealed that the severity of VIDD is dependent on the mode of MV and the levels of spontaneous diaphragm activity. By using the assist-control mode or by allowing intermittent spontaneous breathing during prolonged CMV, the severity of VIDD is mitigated.6 ,7 Prolonged CMV in animals, however, results in decreased isometric force and endurance, muscle fibre atrophy, morphological anomalies of the sarcomere and various mitochondrial-specific dysfunctions.8–17 These pathologies have been attributed to the joint effects of increased protein degradation and decreased protein synthesis.18–21
In brain-dead human organ donors undergoing prolonged MV, several authors have reported the development of diaphragm fibre atrophy, sarcomeric injuries and mitochondrial dysfunction.18 ,19 ,21 ,22 Despite this recent progress in human studies of VIDD, little information is as yet available regarding the effects of prolonged CMV on diaphragm muscle fibre contractile performance. Although it has been reported that significantly lower airway occlusion pressure values are generated in response to phrenic nerve stimulation in intensive care patients undergoing long-term MV (>5 days) as compared with those undergoing short-term MV (0.5 h),18 impaired occlusion pressure merely hints at diminished diaphragm contractility in response to prolonged ventilation.
Similarly, in vitro measurements of isometric force generation of permeabilised single muscle fibre samples from the diaphragms of brain-dead organ donors who had been mechanically ventilated for an average of 26 h were compared with those of control subjects who had been mechanically ventilated for less than 2 h23 and no significant differences were observed between the two groups. But all these indicate that 26 h of CMV is not a sufficient period for sarcomeric contractile dysfunction to develop in humans. The question of whether longer periods of CMV might influence contractility remains untested. Further experimentation is necessary before definitive conclusions can be made, so the first objective of this study is to test the hypothesis that human diaphragmatic sarcomere contractility is negatively affected by CMV administration for periods longer than 26 h. To test this, we used atomic force cantilevers to compare active and passive force generation of diaphragm myofibrils in samples obtained from brain-dead organ donors undergoing prolonged CMV and control subjects who had experienced short periods of MV during cardiac surgery. While active force generation is a result of myofibrillar contraction upon activation, passive force generation is directly associated with the stiffness of titin molecules, which are of special importance for myofibrillar structure and stability. An isolated myofibril is the smallest muscle preparation that has an intact three-dimensional lattice structure, with all relevant proteins present and without the confounding effects that arise from the presence of other intracellular structures. It is also experimentally advantageous to use myofibrils instead of entire muscle fibres because they are of considerably smaller diameter with faster diffusion rates. This prevents the generation of activation gradients during force development and relaxation and allows for clear evaluation of the kinds of potential damage that might unexpectedly affect force development in an intact muscle fibre preparation.
The ability of skeletal muscle fibres to generate active or passive force is determined by levels and post-translational modifications of contractile proteins. While little general information is as yet available on the human diaphragm with regard to fluctuations in contractile protein levels in response to prolonged CMV, significant decreases in myosin heavy chain (MyHC) and actin levels have been reported in rats24–26 and in brain-dead organ donors.20 ,23 These decreases tend to be attributed to increased protein degradation.18–21
To the best of our knowledge, no existing studies have addressed the effects of prolonged MV on any other contractile proteins although fluctuations in myosin light chain (MyLC), troponin, tropomyosin and titin levels are likely to exert a major impact on force generation. Therefore, the second objective of this study is to test the hypothesis that MyLC, troponin-C, troponin-I and troponin-T, tropomyosin and titin levels are all significantly decreased in the diaphragm in response to prolonged CMV and to link these decreases to activation of several proteolytic pathways.
Methods
Experimental subjects
All protocols were approved by the appropriate Ethics Committees of the McGill University Health Centre. All biopsies were performed after written informed consent had been obtained. Full-thickness diaphragm biopsies from 13 normal pulmonary function subjects who were undergoing cardiac surgery (aortic or mitral valve replacement, coronary artery bypass graft) constituted the control group. Biopsies were obtained 30 min after MV was initiated and prior to surgery. Full-thickness diaphragm biopsies from 12 brain-dead organ donors undergoing prolonged CMV constituted the CMV group. Biopsies were obtained prior to circulatory arrest or removal of any organ. All biopsies were obtained from the anterior costal diaphragm, lateral to the insertion of the phrenic nerve. Biopsy samples were immediately frozen in liquid nitrogen, and stored at −80°C until used in immunoblotting and mRNA analyses.
Diaphragm myofibrillar contractile performance
Small muscle bundles were dissected out, tied to wooden sticks and chemically permeabilised according to our laboratory's standard procedures27–29 (see online supplementary file). Briefly, muscle samples were incubated in rigour solution (pH=7.0) for approximately 4 h, after which they were transferred to a rigour:glycerol (50:50) solution for 15 h containing complete protease inhibitor cocktail and then stored at −20°C for at least 7 days. A small piece of muscle (∼1 mm3) was homogenised in rigour solution to isolate myofibrils. Myofibrils were then transferred into a temperature-controlled chamber (15°C) on the stage of an inverted microscope and attached at one end to an atomic force microscope cantilever and at the other to a glass microneedle connected to a piezoelectric motor (see online supplementary file). A multichannel perfusion system connected to a double-barrelled pipette was used to change solutions surrounding the myofibrils to those containing high (pCa2+=4.5) or low (pCa2+=9.0) Ca2+concentrations. When a myofibril contracts, the degree of deflection of the cantilever is detected by a laser beam that is shone onto the back of the cantilever. When the laser light reflects, it is collected and quantified by a specialised photodetection system. Since the stiffness (K) of the cantilever is known, force (F) can be calculated based on its displacement (Δd) during myofibril contractions (F=K×Δd).
Supplemental material
Three procedures were performed on each myofibril:
Active isometric force generation: Myofibrils were isometrically activated for 20 s at an average sarcomere length of 2.4 μm. Maximal force was measured for each contraction.
Rate of force redevelopment: To evaluate cross-bridge kinetics, a slack test to disengage force-generating cross-bridges from actin filaments was performed.30 Myofibrils were activated as they were in the first procedure, but once they achieved maximal force they were shortened (amplitude 30% of sarcomere length; speed of 10 µm/s). Force declined, and then rapidly redeveloped to reach a new steady state. This shortening procedure was performed at varying sarcomere lengths to evaluate cross-bridge kinetics with different degrees of myosin–actin overlap. For each contraction, rates of force development (Kact) and redevelopment (Ktr) were analysed using a double-exponential equation (a×(1−exp(−K×t)−exp (−l×t))+b), and the fast phase rate of relaxation (Krel) was analysed using a single-exponential equation (a×exp (−K×(t−c))+b). For both equations, t is the time, K the rate constant of force development, a the amplitude of the exponential, and b and c are constants.
Passive force generation: Myofibrils were passively stretched from a sarcomere length of 2.2–3.0 µm in 0.2 µm increments in a low Ca2+ concentration buffer (pCa2+=9.0). In total, each myofibril underwent five to eight contractions, including isometric contractions and shortening contractions at different sarcomere lengths, and was passively stretched 1–2 times. Individual values were recorded for statistical comparisons between groups.
RNA extraction
Total RNA was extracted from biopsy samples using a commercial kit. mRNA expressions of four E3 ligases involved in skeletal muscle protein ubiquitination (FBOX32 (Atrogin-1), TRIM36 (MuRF1), TRIM32 and FBOX30 (MUSA1)) were measured with real-time PCR and appropriate primers as previously described.31
Immunoblotting
Samples were loaded onto tris-glycine sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis. Proteins were electrophoretically transferred onto polyvinylidene difluoride membranes, blocked with non-fat dry milk, then incubated overnight with primary antibodies to MyHC-f (fast isoform), MyHC-s (slow isoform), MyLC, α-actin, α-actinin, troponin-C, troponin-I, troponin-T, tropomyosin, LC3B (autophagy marker) and β-tubulin. Proteins were detected using a commercial kit and optical densities (OD) were measured as previously described.31 For a given sample, each protein level was expressed as a ratio of that protein to β-tubulin.
Detection of titin and nebulin proteins
Titin and nebulin isoforms were detected using SDS–agarose electrophoresis as previously described.32 ,33 Briefly, biopsy samples were pulverised using a mortar and pestle and the yield was solubilised in equal volumes of urea buffer (see online supplementary file). Gels (1% agarose) were run for 3 h 20 min at 40°C and were stained using a Silver Stain Plus kit and digitally photographed. OD were measured and expressed as fold change from control values.
20S proteasome activity assay
Frozen biopsy samples were homogenised in homogenisation buffer and centrifuged at 5000 rpm. Supernatants were incubated in a special buffer containing luminogenic Suc-LLVY-aminoluciferin for 30 min at 37°C in the presence and absence of the proteasome inhibitor lactocystin. Substrate cleavage generates a ‘glow-type’ luminescent signal produced by the luciferase reaction; the signal is proportional to proteasome activity. Signals were calibrated using pure 20S proteasome protein and were normalised per microgram muscle protein per min.
Statistical analysis
Continuous measurements that did not follow a normal distribution were compared using a Mann–Whitney test where statistical significance is set at p<0.05. Data are presented using box-and-whisker plots. Continuous measurements that did follow a normal distribution were compared with group t tests and are presented as means±SEM. Statistical analyses were performed with SigmaStat software (Systat Software, Chicago, Illinois, USA). Because the supply of biopsy material was limited, certain analyses could not be performed for all subjects so the number of subjects for each variable is specifically indicated in each figure legend.
For detailed descriptions of all experimental methods, materials and models, see online supplementary file.
Results
Demographic data, duration of CMV, reason for surgery or cause of brain death, relevant medical histories, ventilator settings, arterial blood gases, vital signs and other clinical data for control and CMV groups are listed in tables 1 and 2, and in the online supplementary file. There were no significant differences in age (65.5±3.6 vs 58.0±2.9 years) or body mass index (29.1±2.4 vs 28.8±1.3 kg/m2) between the two groups (table 1). The control group consisted of five females and eight males, while the CMV group consisted of four females and eight males. The duration of MV in the control group was 2–4 h. The duration of MV in the CMV group was 47.6±5.0 h. Systolic blood pressure was the only vital sign that was significantly higher in the CMV group (table 2).
Demographic data, duration of mechanical ventilation, reason for surgery/cause of brain death and relevant medical history for subject diaphragm biopsies in control and case groups
Summary of ventilator settings, arterial blood gases and vital signs for control and CMV groups
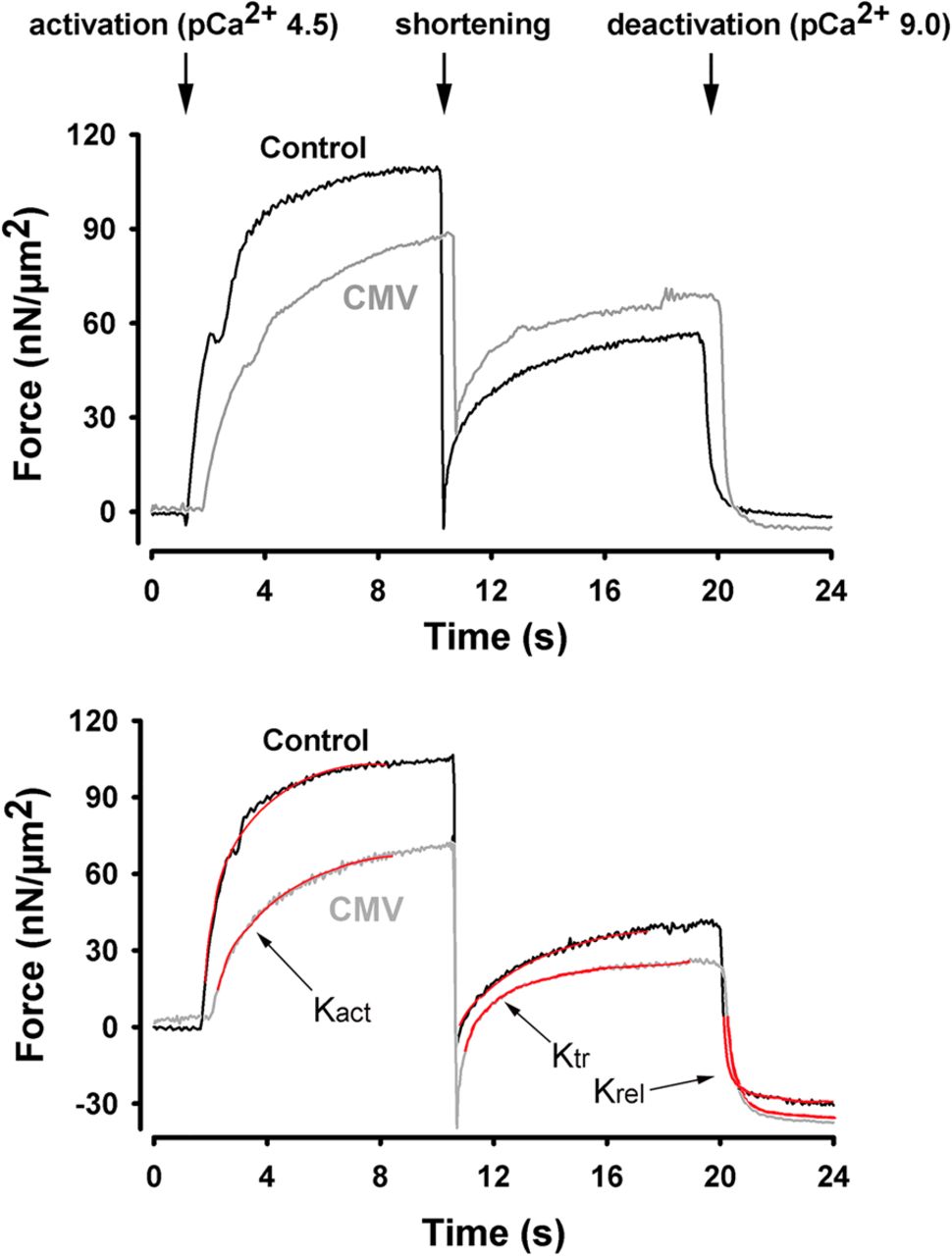
Figure 1A shows typical contractions produced by myofibrils isolated from control and CMV subjects. The active myofibrillar force value in the representative CMV sample was markedly lower than that in the representative control sample, both in terms of absolute value and in terms of specific force, which is force normalised to cross-sectional area (figure 1A, B). For all subjects, specific force generated by myofibrils from the CMV group was significantly lower than that generated by the control group (figure 1C, D, p<0.05).

Active isometric force generation. (A and B) Representative plots of absolute (A) and specific (B) force generation by diaphragm myofibrils isolated from a control subject and a brain-dead organ donor (CMV group) in response to exposure to high Ca2+ concentration buffer (activation) followed by exposure to low Ca2+ concentration (deactivation). Myofibrillar force was measured with an atomic force cantilever at a sarcomere length of 2.2 μm. (C and D) Absolute (C) and specific (D) force generation by diaphragm myofibrils isolated from control and CMV groups. *p<0.05, as compared with the control group. CMV, controlled mechanical ventilation.
To determine myosin cross-bridge kinetics, we used a modified protocol to measure rates of force development and redevelopment in response to fast shortening of fully activated myofibrils.28 ,29 Rates of force development (Kact) and redevelopment (Ktr) during the initial activation were lower in the CMV myofibrils, independent of sarcomere length (figure 2). This finding suggests that the rates at which myosin–actin cross-bridge formations occur during activation or during redevelopment of force under full activation (avoiding Ca2+ kinetic effects) are significantly impaired in the CMV group (table 3). It should be emphasised that relaxation (Krel) rates remained unchanged, indicating that prolonged CMV does not adversely affect myosin–actin dissociation.

Cross-bridge kinetics. Representative plots of force generation by diaphragm myofibrils isolated from a control subject and a brain-dead organ donor (CMV group) in response to exposure to high Ca2+ concentration buffer (activation). Once maximal force was reached, myofibrils were exposed to a shortening procedure (amplitude 30% of sarcomere length; speed 10 µm/s) during which force declined then rapidly redeveloped to reach a new steady state. Shortening procedure was performed at varying sarcomere lengths. Experiments were conducted at sarcomere lengths of 2.2 μm (top panel) and 2.8 μm (bottom panel). Red lines indicate how Kact, Ktr and Kre were determined. CMV, controlled mechanical ventilation.
Summary of changes in diaphragm myofibrillar force development/relaxation in control and CMV groups
To assess passive force generation by diaphragm myofibrils, incremental stretches were applied to rested myofibrils bathed in a low Ca2+ concentration buffer (pCa2+=9.0) (figure 3A). Specific forces generated during passive stretching were significantly lower in myofibrils from the CMV group, as compared with that of the control group (figure 3B).
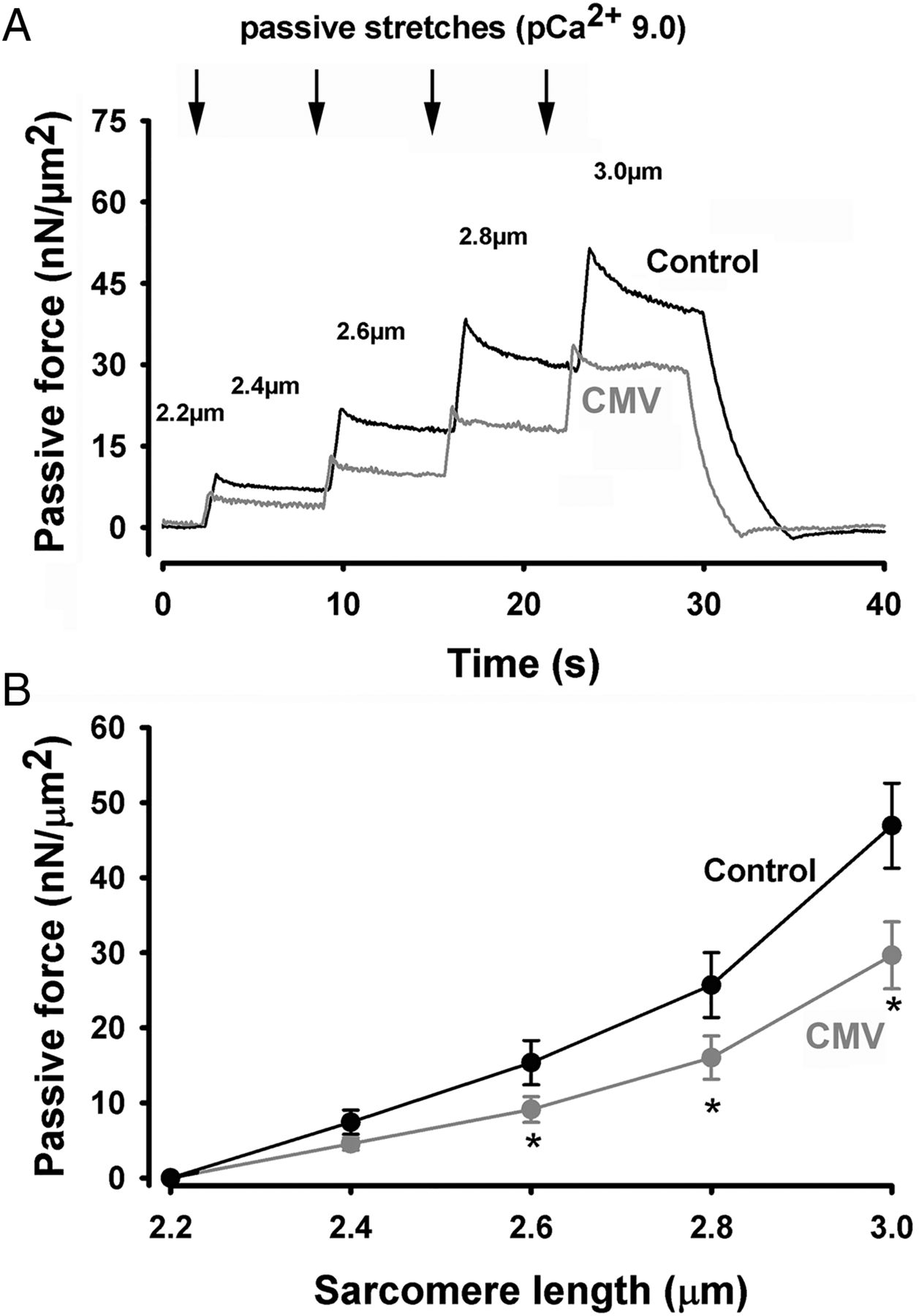
Passive force generation. (A) Representative plot of passive force generation by diaphragm myofibrils isolated from a control subject and a brain-dead organ donor. Myofibrils were progressively stretched at different sarcomere lengths (indicated by arrows and numbers) at increments of 0.2 μm. (B) Passive force generation by diaphragm myofibrils isolated from control (n=10) and CMV (n=8) groups at different sarcomere lengths. Values are means±SEM. *p<0.05, as compared with the control group. CMV, controlled mechanical ventilation.
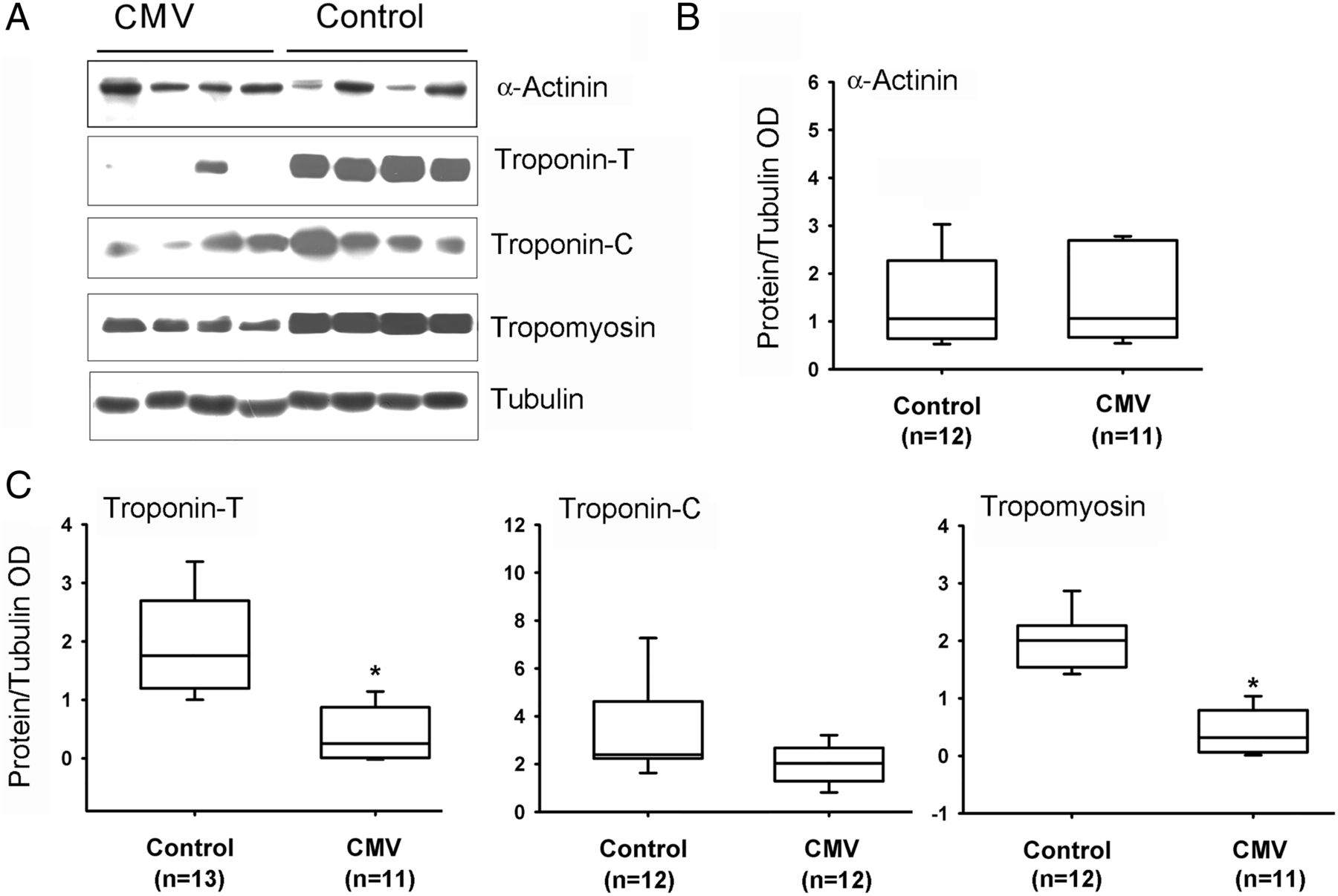
Slow MyHC isoform protein levels were significantly lower in CMV subjects as compared with the control group (figure 4). No differences were observed between the two groups in levels of fast MyHC or α-actin (figure 4). Fast and slow isoforms of MyLC 1-3 and 2 proteins were detected with four different antibodies (see online supplementary file). Both isoforms of MyLC1-3 were significantly lower in the CMV group (figure 5). No significant differences in MyLC-2 (figure 5), α-actinin or troponin-C were observed (figure 6). Troponin-T, tropomyosin (figure 6) and troponin-I (figure 7) were significantly lower in the CMV group. Titin levels were significantly lower in the diaphragms of the CMV group but no significant differences in nebulin levels were observed (figure 8).
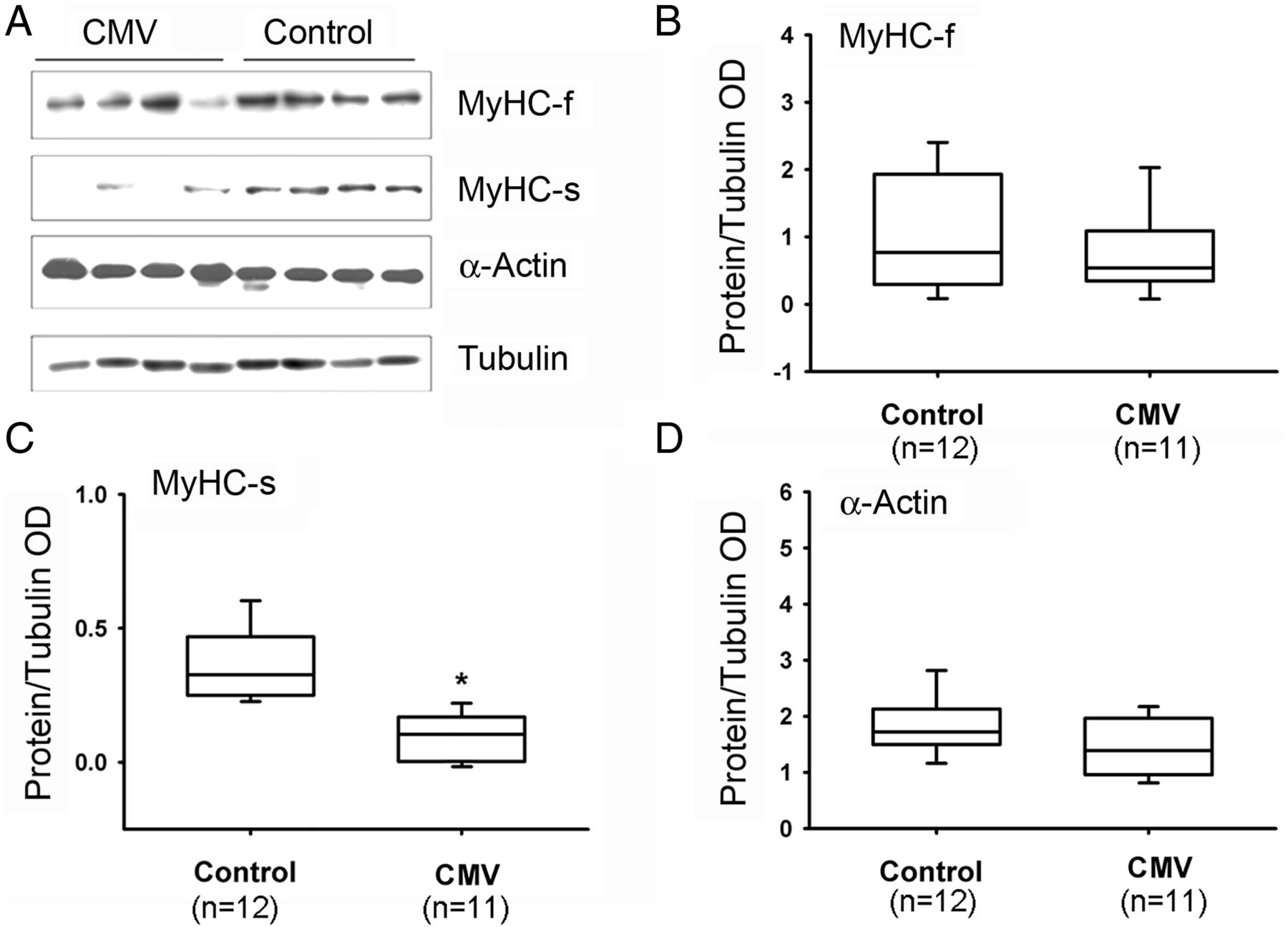
Protein expressions of MyHC and actin. (A) Representative protein immunoblots of fast (f) and slow (s) isoforms of MyHC and α-actin in the diaphragms of control and CMV groups. (B–D) Protein levels of MyHC-f, MyHC-s and α-actin in the diaphragms of control and CMV groups. *p<0.05, as compared with the control group. CMV, controlled mechanical ventilation; MyHC, myosin heavy chain; OD, optical densities.

Protein expression of MyLC. (A) Representative protein immunoblots of the fast (f) and slow (s) isoforms of MyLC1 and MyLC2 in the diaphragms of control and CMV groups. (B–D) Protein levels of MyLC1-f, MyLC1-s and MyLC2 in the diaphragms of control and CMV groups. *p<0.05, as compared with the control group. CMV, controlled mechanical ventilation; MyLC, myosin light chain; OD, optical densities.

Protein expressions of actinin, tropomyosin, troponin-T and troponin-C. (A) Representative protein immunoblots of α-actinin, troponin-T, troponin-C and tropomyosin in the diaphragms of control and CMV groups. (B–D) Protein levels of α-actinin, troponin-T, troponin-C and tropomyosin in control and CMV groups. *p<0.05, as compared with the control group. CMV, controlled mechanical ventilation; OD, optical densities.

Protein expression of troponin-I. (A) Representative protein immunoblots of slow (s) and fast (f) isoforms of troponin-I and tubulin in the diaphragms of control and CMV groups. (B) Troponin-I-s and troponin-I-f protein levels in control and CMV groups. *p<0.05, as compared with the control group. CMV, controlled mechanical ventilation.

Protein expressions of titin and nebulin. (A) Representative protein gel blots of titin and nebulin proteins in the diaphragms of control and CMV groups. (B) Titin and nebulin protein levels in control and CMV groups. Expressed as fold change from control. *p<0.05, as compared with the control group. CMV, controlled mechanical ventilation; OD, optical densities.
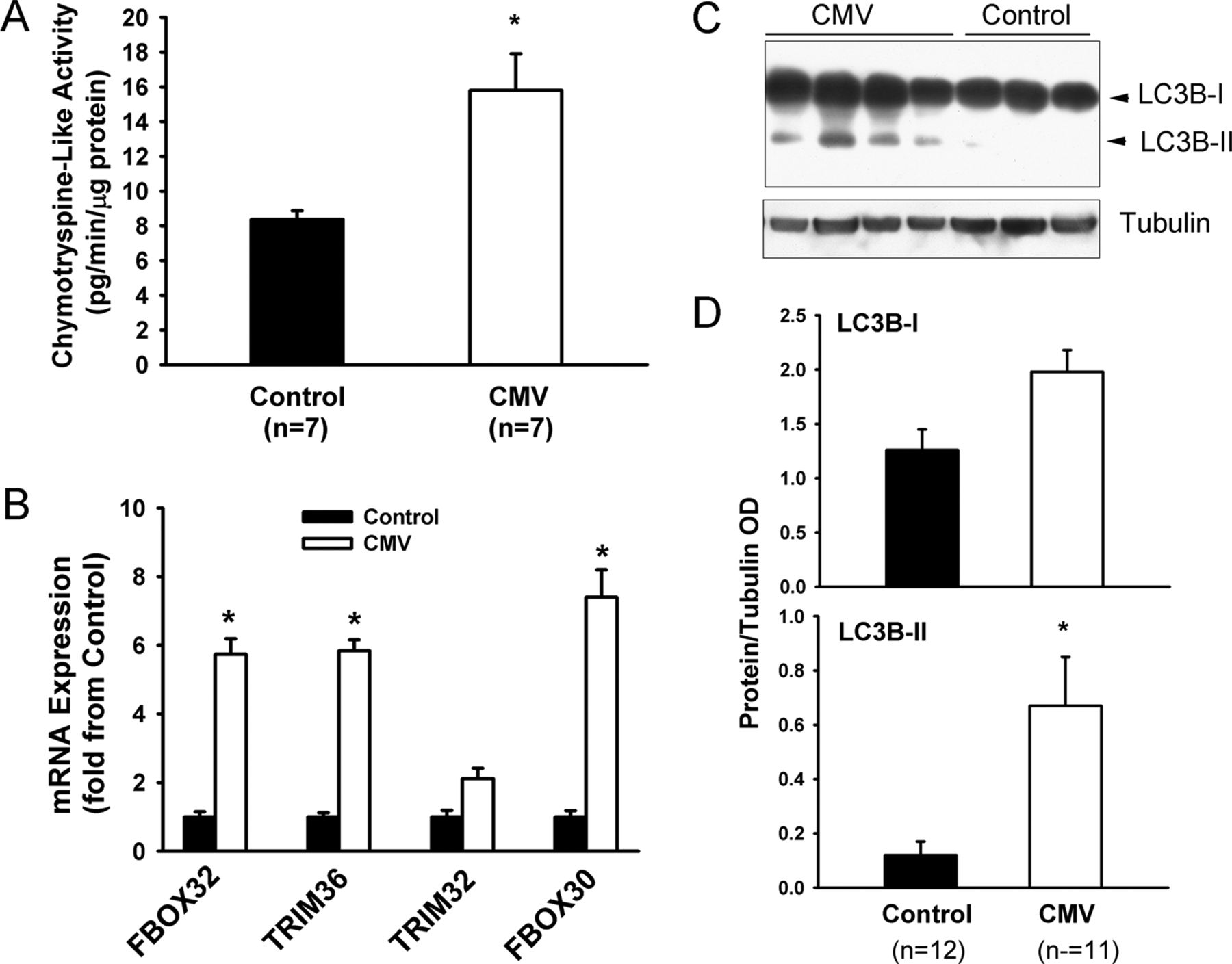
Compared with the control group, chymotrypsine-like 20S proteasome activity significantly increased in the diaphragms of the CMV group (figure 9A). mRNA expressions of three E3 ligases involved in skeletal muscle protein ubiquitination (FBOX32, TRIM36 and FBOX30) significantly increased in the CMV group (figure 9B). Levels of the lipidated form of LC3B (LC3B-II) significantly increased in the CMV group, indicating that autophagy had been induced (figure 9C, D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Activation of the ubiquitin-26S proteasome and autophagy–lysosome pathways. (A) Chymotrypsine-like activity of the 20S proteasome in the diaphragms of the control and the controlled mechanical ventilation (CMV) groups. Values are means±SEM and are expressed as pg/min/μg total muscle protein. *p<0.05, as compared with the control group. (B) Expression of FBOX32 (Atrogin-1), TRIM36 (MuRF1), TRIM32 and FBOX30 (MUSA1) mRNA levels in diaphragms of the control and the CMV group. Values are means±SEM and are expressed as fold change from the control group. *p<0.05, as compared with the control group. (C) Representative immunoblots of LC3B and tubulin proteins in the diaphragms of control and CMV groups. LC3B-I refers to cytosolic form of LC3B protein. LC3B-II refers to lipidated form of LC3B protein, which is incorporated into autophagosome membranes. (D) Optical densities (OD) of LC3B-I and LC3B-II detected in the diaphragms of the control and the CMV groups. Values are means±SEM and are expressed as a ratio of OD of protein to OD of tubulin. *p<0.05, compared with the control subjects.
Discussion
In this study, we investigated the relationship between myofibrillar contractility, myofilament protein level and prolonged CMV in the human diaphragm. Our results indicate that prolonged CMV is associated with the following changes: (1) reduced maximal active and passive specific force generation in myofibrils; (2) reduced force redevelopment during activation and in response to imposed shortening; (3) reduced levels of the myofilament proteins MyHC (slow), MyLC 1, tropomyosin, troponin-I, troponin-T and titin; (4) induction of 20S proteasome activity and mRNA expressions of E3 ubiquitin ligases; and (5) induction of the autophagy–lysosome pathway.
CMV-induced impairment of diaphragm contractility
Several animal studies have confirmed that prolonged CMV is associated with a significant decline in diaphragm contractile performance.6 ,10 ,12 ,17 ,34–36 To the best of our knowledge, no studies have been published that document the same effect in non-critically ill humans.37 As an indirect indicator of diaphragm strength, Jaber et al18 measured tracheal occlusion pressure in response to bilateral phrenic nerve stimulation in intensive care patients undergoing prolonged MV. They reported a progressive decline in pressure with a mean reduction of 32% after 6 days. However, the observed decline in occlusion pressure is just as likely to have been caused by any number of other factors, such as defective phrenic nerve conduction or neuromuscular transmission, impaired excitation–contraction coupling within diaphragm muscle fibres and/or actin–myosin interaction abnormalities.
Hooijman et al23 recently performed an elegant study in which they compared contractile performance (maximum isometric force, cross-bridge kinetics and Ca2+ sensitivity) of permeabilised single muscle fibres derived from diaphragms of brain-dead organ donors undergoing an average of 26 h of CMV to those derived from control subjects exposed to 2 h of MV while undergoing lung cancer surgery. They failed to detect any differences between the two groups and concluded that 26 h of CMV in humans does not affect contractile performance of sarcomeres. The discrepancy between their results and ours may be due to several factors relating to differences in the methods and preparations used for measuring contractile performance measurements in each study. First, we used individual myofibrils isolated from muscle samples, whereas Hooijman et al used whole fibres. The isolated myofibril preparation allows for more accurate measurements of protein contractile activity by avoiding the kinds of artefactual effects of non-contractile intracellular elements that occur in single fibre preparations. Second, the duration and mode of ventilation applied to the CMV group were different in each study. In the Hooijman et al study, the average duration of MV was 26 h, whereas in our study it was 47 h. In fact, they suggested that this relatively short duration of CMV might explain the lack of diaphragm muscle atrophy and relatively normal (or event elevated) MyHC content that was observed in their CMV group.18 We should emphasise, however, that the duration of MV indicated in various studies using brain-dead organ donors is primarily based on clinical information that was recorded after the subject was declared brain dead and therefore may not reflect the actual total duration of MV that these subjects experienced. Third, differences in patient characteristics (age, cause of death, past medical history, medication use and nutritional status) can result in varying conclusions being made about contractile performance. Fourth, the control groups in each study are significantly different in terms of smoking history and reason for thoracic surgery. In our study, the control group consists of patients with cardiac valvular disease who were undergoing surgery for valve replacements. These patients had no history or current manifestations of skeletal muscle dysfunction or atrophy. In addition, we avoided the confounding effects of prolonged anaesthesia by obtaining diaphragm biopsies within very short time periods following initiation of anaesthesia. In the Hooijman et al study, the control group consisted of current or ex-smokers who were undergoing thoracic surgery for resection of lung nodules. Some of them might have already had abnormally low diaphragm sarcomeric contractile function as a consequence of chronic smoking and/or lung cancer.
Mechanisms of impaired diaphragm myofibrillar contractility
The molecular mechanisms that underpin the impaired active force generation of diaphragm myofibrils isolated from the CMV group have not yet been identified. Active force generation is determined by the proportion of cross-bridges strongly bound to actin in a given time period during activation, or the force generated by individual cross-bridges while attached to actin.38 Measurements of the rates of force development (Kact) and force redevelopment following imposed shortening (Ktr) during active force generation provide relevant information regarding the rates of cross-bridge attachment and detachment, which are, in turn, reflective of the fraction of strongly bound cross-bridges. Analyses of these rates have commonly been used to detect changes in cross-bridge kinetics in several different diseases that affect muscles.30 ,39
In this study, we observed significant decreases in Kact and Ktr in myofibrils from the CMV group (table 3), suggesting a slowing down of cross-bridge kinetics and a reduction in the proportion of cross-bridges strongly bound to actin. We also found changes in the content of slow MyHC, known to be a major determinant of maximum specific force generation in diaphragm muscle fibres.40 In comparison, Hooijman et al,23 who found no differences in MyHC levels between brain-dead organ donors and control subjects, used a shorter duration of MV than what was used by either Levine et al or by our group. With current technology, we cannot measure a relationship between impairment of force and changes in MyHC content in individual myofibrils, and it is possible that myofibrils with the slow MyHC isoform were more affected than myofibrils with the fast MyHC isoform. Such a relationship should be investigated in the near future although our overall results would still indicate a decrease in diaphragm contractility.
One likely mechanism for the decline in active force generation in response to CMV relates to decreases in the levels of several regulatory contractile proteins including MyLC, troponin-I, troponin-T and tropomyosin. We detected fast and slow MyLC1 and MyLC2 isoforms by immunoblotting with two separate antibodies. This revealed significantly lower levels of MyLC1 in the CMV group, but no difference in MyLC2 levels between the two groups (figure 5). These results suggest that MyLC1 is particularly sensitive to the effects of prolonged MV as compared with MyLC2. Tropomyosin, which is a dimeric coiled-coil acting-binding protein that acts as a ‘gatekeeper’ of thin filament Ca2+ activation and thereby regulates active force generation in striated sarcomeres, was also significantly decreased in the diaphragms of the CMV group, as were the levels of troponin-T and troponin-I (figures 6 and 7). Troponin-T is a protein that links the troponin complex to tropomyosin and actin while troponin-I is a protein that holds other troponins together and onto actin by directly binding to actin. Troponin-I exerts a negative influence on the binding of myosin and on activation of actomyosin ATPase activity.41
We also observed that passive force generation of diaphragm myofibrils was significantly lower at a given sarcomere length in response to CMV. van Hees et al25 reported significant impairment of passive force generation by diaphragm fibres in response to MV in rats. Since the protein titin is the most important determinant of passive elastic properties of skeletal muscle,42 we suggest that reduction in passive force generation in the CMV group might be due to reduction in titin levels (figure 8). We do not exclude, however, the possibility that alterations in titin phosphorylation25 might have contributed to lower passive force generation in the CMV group. It should be noted that decreased titin levels may not just affect passive force generation but may also affect sarcomere stability and active force generation. Since titin is anchored to thick filaments, a reduction in titin would significantly increase the instability of sarcomeres.43
Significant decreases in contractile protein levels in the CMV group are likely due to increased degradation and/or decreased synthesis. Prolonged MV has been linked to significant induction of four proteolytic pathways (ubiquitin-26S proteasome, calpain, caspase-3 and autophagy–lysosome pathways).18–21 ,24 ,44 In this study, we confirmed that the ubiquitin-26S proteasome and autophagy pathways are induced in the CMV group. This observation suggests that increased proteolysis might be the reason for lower contractile protein levels. It is unclear from this study, however, whether decreased synthesis might also have contributed to the decline. We would like to emphasise that mechanisms other than those that relate to decreased myofilament protein levels might have contributed to the impairment of diaphragm myofibrillar contractility in the CMV group. These mechanisms include ultrastructural sarcomere injury,18 decreased mitochondrial biogenesis and dysfunction,22 lipid accumulation22 and oxidative injury.20 ,21 ,45
Limitations of the study
One limitation of our study relates to the use of brain-dead organ donors as a model to study the effects of prolonged MV on diaphragm function in humans and caution should be exercised when extrapolating findings obtained from this model to critically ill patients undergoing prolonged MV.18–23 Currently, the best available model is that of Hooijman et al37 in which diaphragm and abdominal muscle biopsies are obtained from critically ill patients undergoing prolonged MV. The weakness of the brain-dead organ donor model is related to several factors, among which is the mode of ventilation. In brain-dead organ donors, CMV is the standard mode of ventilation. Pressure-support ventilation or modes that allow for spontaneous diaphragm activity are used for critically ill patients and likely elicit milder forms of VIDD than those described in brain-dead organ donors. Brain death may also trigger nutrition-related biochemical defects and hormonal abnormalities that may influence skeletal muscle structure and function.46 ,47 Although the contribution of these abnormalities to the impairment of contractile performance or reductions in protein level in the CMV group cannot be excluded in our study, previous reports indicate that muscle fibre atrophy and induction of proteolysis via the ubiquitin-26S proteasome and autophagy pathways in brain-dead organ donors undergoing prolonged CMV are limited to the diaphragm.19 ,21 ,22 In this study, therefore, unloading and disuse are very likely to have been crucial determinants in the development of impaired contractility and decreased protein expression.
Acknowledgments
The authors are grateful to Ms Anne Gatensby for editing the manuscript.
References
Footnotes
Contributors SNAH: Design of study, data analysis, manuscript writing and editing. AC: Data collection and analysis. CG: Data collection and analysis. AAK: Data collection, analysis, manuscript writing and editing. FdSL: Data collection and analysis. BJP: Data collection and analysis. MM: Data collection, analysis, writing of manuscript. NM: Data collection and analysis. BdV: Data collection. PG: Study design, data collection, writing of manuscript. DR: Study design, data collection and analysis, writing and editing manuscript.
Funding This study was funded by grants from the Canadian Institutes of Health Research and Natural Sciences and Engineering Research Council of Canada. ASC is a recipient of a post-doctoral fellowship (2013/07104-6) from FAPESP, São Paulo, Brazil. FdSL is a recipient of a scholarship from CNPq (Government of Brazil).
Competing interests None declared.
Patient consent Obtained.
Ethics approval McGill University Health Centre.
Provenance and peer review Not commissioned; externally peer reviewed.