Article Text

Abstract
Background: Obstructive sleep apnoea (OSA) elicits a number of cardiovascular perturbations that could lead acutely or chronically to increased ventricular ectopy in patients with heart failure (HF). We tested the hypothesis that treatment of OSA with continuous positive airway pressure (CPAP) in patients with HF would reduce the frequency of ventricular premature beats (VPBs) during sleep in association with reduced sympathetic nervous system activity.
Methods: Following optimisation of medical treatment, 18 HF patients with OSA and >10 VPBs per hour of sleep were randomised to a control group (n = 8) or a treatment group who received CPAP (n = 10). The frequency of VPBs and urinary norepinephrine (noradrenaline) concentrations during total sleep time were determined at baseline and after 1 month.
Results: Control patients did not experience any significant changes in apnoea-hypopnoea index (AHI), mean nocturnal O2 saturation, or the frequency of VPBs. In contrast, there was a significant reduction in AHI (p<0.001), an increase in minimum O2 saturation (p = 0.05), a reduction in urinary norepinephrine concentrations (p = 0.009), and a 58% reduction in the frequency of VPBs during total sleep (from mean (SE) 170 (65) to 70 (28) per hour, p = 0.011) after 1 month of CPAP treatment.
Conclusions: In patients with HF, treatment of co-existing OSA by CPAP reduces the frequency of VPBs during sleep. These data suggest that reductions in VPBs and other ventricular arrhythmias through treatment of OSA might improve the prognosis in patients with HF.
- AHI, apnoea-hypopnoea index
- CPAP, continuous positive airways pressure
- HF, heart failure
- LVEF, left ventricular ejection fraction
- OSA, obstructive sleep apnoea
- VPB, ventricular premature beat
- heart failure
- sleep apnoea
- urinary norepinephrine
- ventricular ectopy
Statistics from Altmetric.com
- AHI, apnoea-hypopnoea index
- CPAP, continuous positive airways pressure
- HF, heart failure
- LVEF, left ventricular ejection fraction
- OSA, obstructive sleep apnoea
- VPB, ventricular premature beat
In normal healthy subjects asymptomatic ventricular premature beats (VPBs) are considered benign1 and can be triggered by increased sympathetic outflow to the ventricles.2 The frequency of VPBs normally falls during non-rapid eye movement (NREM) sleep, which comprises approximately 85% of total sleep time. This decline parallels the decrease in sympathetic outflow and the increase in vagal outflow to the heart that accompanies the transition from wakefulness to NREM sleep.3 Conversely, the frequency of VPBs tends to rise just before and after waking, a time when cardiac sympathetic tone increases.4,5 Several other factors stimulate ventricular ectopy including myocardial stretch due to both transient and sustained dilatation, leading to mechano-electrical dissociation.6,7 Patients with heart failure (HF) have an increased risk of sudden death, often in relation to ventricular arrhythmias.8 While administration of antiarrhythmic drugs specifically designed to reduce ventricular ectopy does not improve survival in patients with HF,9 β blockers—which attenuate the impact of cardiac sympathetic stimulation and reduce myocardial ischaemia—also reduce the frequency of both ventricular arrhythmias and sudden death.10,11
An association between atrial arrhythmias and obstructive sleep apnoea (OSA) has been described.12 The recent report of an excess of sudden deaths during sleep between midnight and 06.00 hours in patients with OSA also suggests that OSA may trigger lethal nocturnal ventricular arrhythmias.13 In subjects with OSA, recurrent nocturnal apnoeas, hypoxia and arousals from sleep trigger acute increases in sympathetic outflow during sleep14 and, in patients with coronary artery disease, can trigger nocturnal ischaemia and angina.15 The co-existence of OSA in approximately one third of patients with HF may therefore further augment sympathetic outflow during sleep and put these patients at risk for nocturnal myocardial ischaemia15 and ventricular arrhythmias.16
Treatment of OSA by continuous positive airway pressure (CPAP) in patients with normal ventricular function has been shown to prevent apnoea related surges in muscle sympathetic activity and blood pressure during sleep.17 In patients with HF the treatment of co-existing OSA reduces both nocturnal blood pressure and heart rate.18 Furthermore, randomised trials involving patients with HF have shown that treatment of coexisting OSA by CPAP reduces nocturnal urinary catecholamines17 and daytime muscle sympathetic nerve activity,19 lowers daytime blood pressure,19,20 and improves daytime left ventricular ejection fraction (LVEF).20,21 However, the effects of CPAP on ventricular ectopy have not been examined in the context of a randomised trial. We therefore conducted a randomised controlled trial of treatment of OSA by CPAP in patients with HF to test the hypothesis that abolishing OSA would reduce the frequency of ventricular ectopy and urinary norepinephrine (noradrenaline) excretion during sleep.
METHODS
Subjects
Study subjects were recruited from the heart failure clinics of the Mount Sinai and Toronto General Hospitals in Toronto. Patients referred to these clinics routinely undergo overnight polysomnography. Study entry criteria were a history of HF of at least 6 months’ duration due to ischaemic or non-ischaemic dilated cardiomyopathy; left ventricular systolic dysfunction as evidenced by an LVEF of ⩽45% at rest by gated radionuclide angiography; New York Heart Association functional class II to IV; the presence of sinus rhythm; stable clinical status for at least 1 month before participation in the trial while on optimal pharmacological treatment at the highest tolerated doses; moderate to severe OSA defined as an apnoea-hypopnoea index (AHI) ⩾20/hour of sleep of which >50% were obstructive; and >10 VPBs/hour of sleep.22 Exclusion criteria were primary valvular heart disease; the presence of a cardiac pacemaker; and unstable angina, myocardial infarction or cardiac surgery within the previous 3 months.
This project was part of a larger research study to examine a number of cardiovascular effects of CPAP in HF patients with OSA.19,20 Hence, patients with ventricular ectopy were identified following the randomisation process. The protocol was approved by the Human Subjects’ Review Committee of the University of Toronto and all patients gave written consent before participation.
Sleep studies
Overnight sleep studies were performed using standard techniques and criteria for scoring sleep stages and arousals.23,24 Thoracoabdominal movements and tidal volume were monitored by a calibrated respiratory inductance plethysmograph (Respitrace, Ambulatory Monitoring Inc, White Plains, NY, USA) and arterial oxyhaemoglobin saturation (Sao2) by a pulse oximeter (Nellcor N200; Nellcor Puritan Bennett Inc, Pleasanton, CA, USA).24 All signals were recorded on a computerised sleep scoring system (Sandman; Tyco Ltd, Ottawa, Canada). Apnoeas were defined as an absence of tidal volume for >10 seconds and were classified as obstructive if there was paradoxical thoracoabdominal motion and as central if there was no thoracoabdominal motion. Hypopnoeas were defined as a reduction in tidal volume of >50% for >10 seconds. Hypopnoeas were classified as obstructive or central in the presence or absence of out of phase thoracoabdominal motion, respectively. Scoring of apnoeas and hypopnoeas was performed by a polysomnographic technician who was blinded to the electrocardiographic (ECG) findings. The AHI per hour of sleep was calculated. The ECG and heart rate were monitored continuously via lead I and sampled at a frequency of 1000 Hz.
Cardiovascular assessment
The ECG tracing during all stages of sleep was inspected for the presence of VPBs. The QRS complexes were scored as VPBs if they were: (1) premature, (2) not preceded by a premature P wave, (3) ⩾0.12 seconds in duration, and (4) of different morphology from those arising from sinus beats. So few couplets and episodes of ventricular tachycardia were present that no analysis of these arrhythmias was performed. Urinary norepinephrine concentrations, corrected for creatinine excretion, were determined from complete 8 hour overnight urine collections obtained during overnight sleep studies as previously described.25
Blood pressure was measured continuously by digital photoplethysmography (Finapres 2300; Ohmeda, Englewood, CO, USA) over a 10 minute period in the morning following the sleep study with the subject awake and supine. Following blood pressure measurements, two dimensional echocardiography was performed on all subjects. Left ventricular end diastolic and end systolic volumes were determined and LVEF was calculated using the modified Simpson’s method26 by a senior ultrasonographer who was unaware of the treatment allocation of the patient or the purpose of the study.
Study protocol
Following the baseline assessment, subjects were randomised to a control group who continued on optimal drug treatment for HF, or to a treatment group who received CPAP in addition. The night after the baseline sleep study patients randomised to this intervention underwent overnight CPAP titration during which pressure was adjusted to abolish apnoeas and hypopnoeas, or to the highest level tolerated. They were then provided with a metered CPAP machine to document hours of use and prescribed its application for >6 hours each night. Baseline studies were replicated after 1 month.
Statistics
All data were acquired and analysed blindly by personnel unaware of treatment allocation. Data were expressed as mean (SE). Statistical analysis was performed using Sigmastat 2.03 (SPSS Inc, Chicago, IL, USA). Baseline characteristics of the control and CPAP treated groups were compared by two tailed unpaired t tests for continuous variables and the Fisher’s exact test for nominal variables. Two way repeated measures analyses of variance followed by Tukey’s test were used to compare within and between group differences in variables obtained 1 month apart. To take into account potential differences in baseline rates of VPBs between the two groups, analysis of covariance (ANCOVA) was also used to analyse time-treatment interactions between baseline and 1 month. p values of <0.05 were considered significant.
RESULTS
Subject characteristics
Figure 1 illustrates the flow of eligible patients through the study. Of the 18 patients identified as having >10 VPBs per hour of total sleep time during baseline studies, 12 had ischaemic cardiomyopathy and six had non-ischaemic dilated cardiomyopathy; eight were randomised to the control group and 10 to the CPAP treatment group. As shown in table 1, there were no significant differences between the two groups with respect to age, sex, body mass index, cause of HF, NYHA class, LVEF, Epworth sleepiness scale, or medication use. The AHI and frequency of arousals from sleep were significantly lower in the group randomised to CPAP than in the control group (table 2). However, at baseline there were no significant differences in the frequency of VPBs between the CPAP and control groups during sleep (fig 2). The frequency of VPBs did not differ significantly between NREM and REM sleep for all subjects (118.4 (32.1)/h v 163.0 (55.5)/h, p = 0.9). There was no significant correlation between VPBs/h and AHI, minimum Sao2, body mass index, age, systolic or diastolic blood pressure, or LVEF.
Mean (SE) baseline characteristics of study subjects
Mean (SE) polysomnographic outcomes
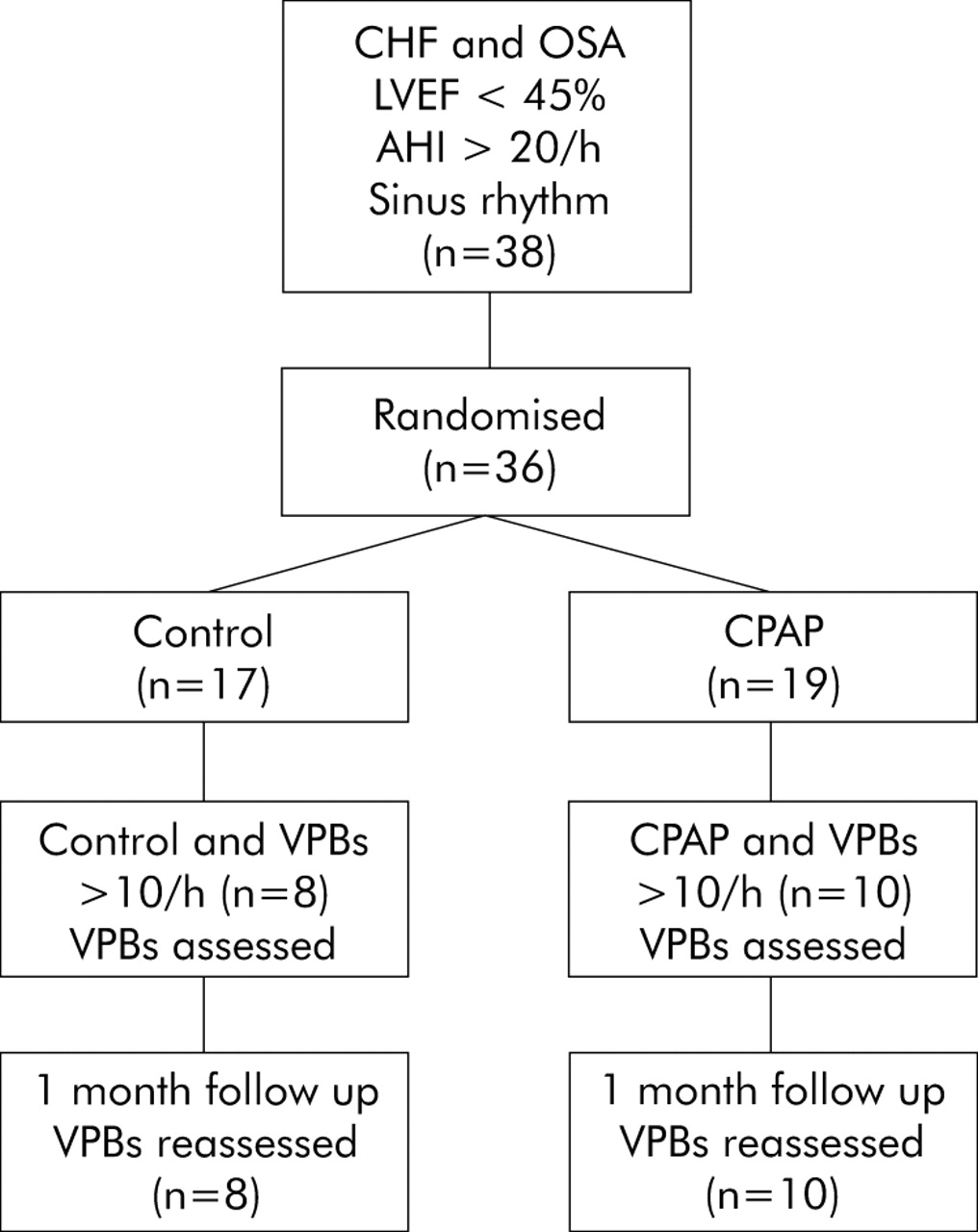
Flow diagram indicating progress of eligible subjects through the study.

{kind=link}
{kind=link}
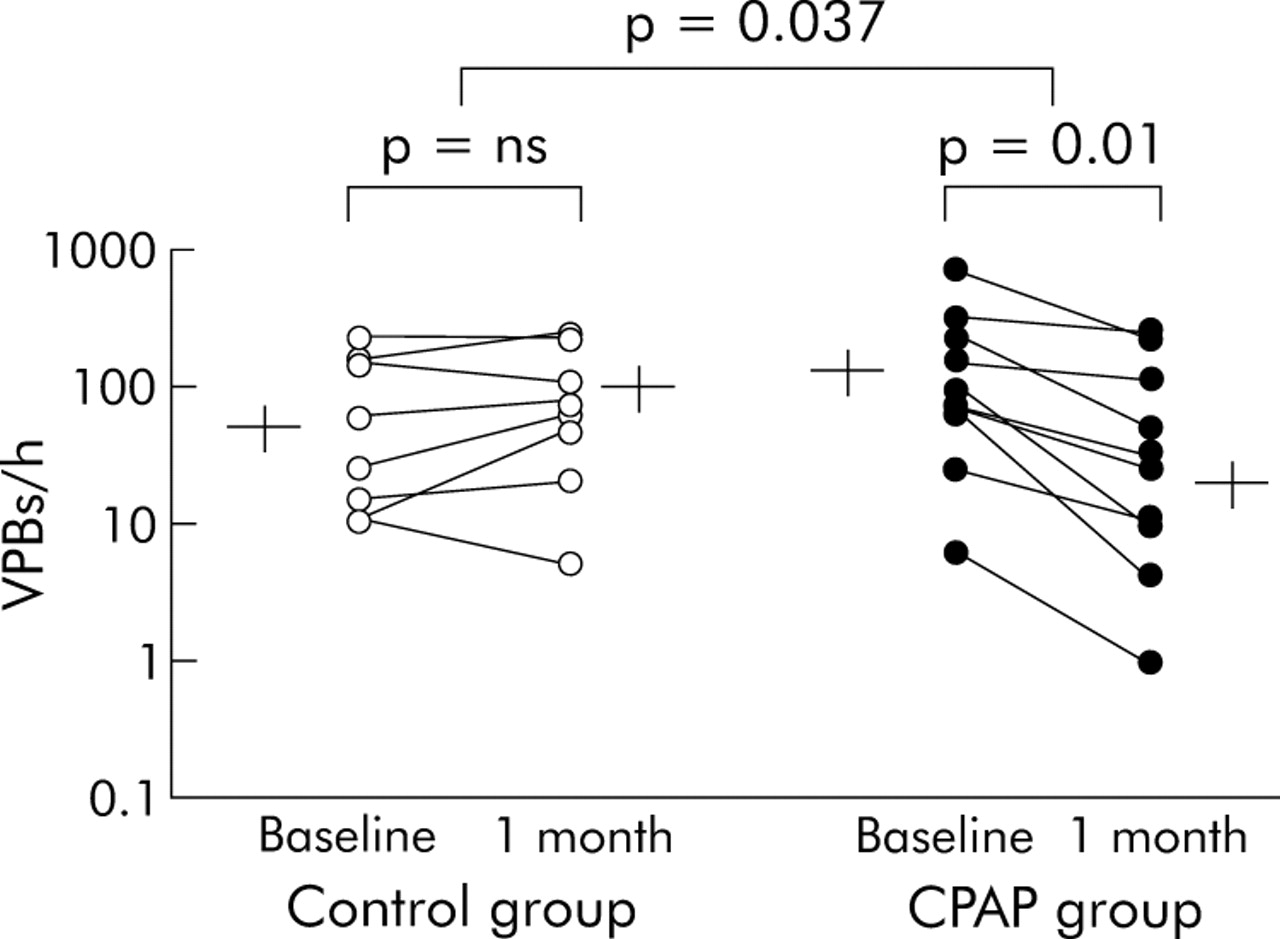
Ventricular premature beats (VPBs) during total sleep in individual subjects with group mean (SE) values at baseline and after 1 month in control subjects (left panel) and CPAP treated subjects (right panel). Note data are plotted on a log scale. There was no change in the frequency of VPBs in the control group (from 84 (31) to 101 (33)/h). In contrast, the frequency of VPBs fell significantly in all subjects receiving CPAP (from 170 (65) to 70 (28)/h, p = 0.011 for within group comparisons, p = 0.037 for between group comparisons, and p = 0.03 for between group comparisons by analysis of covariance).
Effects of CPAP
No changes in medication or body mass index occurred after 1 month in either group (table 1). In the control group there was a significant increase in total sleep time (p = 0.002) and a significant decrease in minimum Sao2 (p = 0.004), but no changes in the AHI, frequency of arousals from sleep, or mean Sao2 (table 2). Patients randomised to CPAP used it for 6.2 (0.5) hours per night at a mean pressure of 8.0 (0.5) cm H2O (range 6–10). CPAP treated subjects experienced significant reductions in AHI (p<0.001), frequency of arousals (p = 0.004), and an increase in minimum Sao2 (p = 0.05), but no change in total sleep time (table 2).
Compared with the control group, daytime systolic blood pressure fell significantly by 4.6 mm Hg in the CPAP treated subjects (p = 0.004 for between group comparison). There were no corresponding changes in diastolic blood pressure or heart rate. There was a significant improvement in LVEF in the CPAP treated group (table 3, p = 0.034 and 0.030 for within and between group comparisons). However, although there were tendencies for reductions in both LV end systolic and end diastolic volumes in the CPAP treated group, these differences were not significant (table 3).
Mean (SE) cardiovascular variables
As shown in fig 2, in the control group the frequency of VPBs during sleep did not change significantly from baseline to 1 month. In contrast, in CPAP treated subjects the frequency of VPBs during total sleep time decreased by 58% (p = 0.011 for within group comparisons, p = 0.037 for between group comparisons, and p = 0.03 for between group comparisons by analysis of covariance). One subject in the CPAP treated group had a much higher rate of VPBs during sleep than the others. Nevertheless, even when data from this subject were excluded so that the mean frequency of VPBs at baseline was more similar to that of the control group, the CPAP induced reduction in the frequency of VPBs for total sleep remained highly significant (111.5 (31.8)/h v 53.6 (25.9)/h, p = 0.002 and p = 0.004 for within and between group comparisons, respectively, and p = 0.006 for between group comparisons by analysis of covariance).
When sleep was subdivided into NREM and REM sleep, there were no significant changes in the frequency of VPBs in the control group during the 1 month trial period in either stage (NREM sleep: from 81 (31) to 100 (32)/h; REM sleep: from 114 (49) to 115 (126)/h). In contrast, in the CPAP treated group the frequency of VPBs fell significantly both during NREM (from 149 (52) to 88 (40)/h, p = 0.013 for within group differences, p = 0.026 for between group differences) and REM sleep (from 202 (294) to 35 (43)/h, p = 0.023 for within group differences).
In association with the reduction in VPB frequency there was also a reduction in the overnight urinary norepinephrine concentration in the CPAP group compared with the control group (table 3, p = 0.009 for between group comparisons).
DISCUSSION
It has long been speculated that OSA can provoke ventricular arrhythmias and that treatment of OSA can reverse them.27 However, there has been no evidence from randomised trials to substantiate these assumptions. Consequently, our data are important because they are the first from a randomised controlled clinical trial to confirm that alleviation of OSA by CPAP during sleep in patients with HF significantly reduces ventricular ectopy during sleep. These observations suggest that OSA is a trigger for ventricular ectopy that is amenable to specific non-pharmacological therapy. There are a number of potential mechanisms through which CPAP could reduce VPB frequency. In patients with HF, acute elimination of OSA by CPAP alleviates apnoea-related hypoxia and arousals from sleep, and lowers left ventricular afterload by reducing blood pressure and abolishing exaggerated negative intrathoracic pressure swings.18,20 Acute application of CPAP to patients with severe HF while awake also reduces their cardiac norepinephrine spillover.28 Recent randomised trials in HF patients with OSA have shown that patients receiving CPAP experienced reductions in nocturnal urinary norepinephrine concentrations21 and a fall in daytime muscle sympathetic nervous activity, in conjunction with a reduction in daytime blood pressure.19 In the present study, CPAP caused marked reductions in the frequency of obstructive apnoeas and hypopnoeas and arousals from sleep that were accompanied by improved nocturnal oxygenation and reduced daytime blood pressure. Thus, one mechanism through which CPAP could reduce the frequency of VPBs is by reducing oxygen demand and increasing oxygen supply.29 This could attenuate VPBs either by preventing ischaemia in patients with ischaemic heart disease or by improving ventricular repolarisation.30 Another potential mechanism is through a reduction in cardiac sympathetic nerve traffic.28 This is supported by the finding of reduced overnight urinary norepinephrine in the CPAP treated group. A third mechanism may involve unloading of the ventricles with the elimination of their transient mechanical distension and a consequent alleviation of electrical-mechanical dissociation.7 The combination of these effects of CPAP could reduce ventricular irritability and decrease ventricular ectopic activity during sleep.
Only one other study has examined the effect of CPAP on ventricular ectopy in HF patients.31 In that study CPAP was applied to a heterogeneous group of HF patients with either central sleep apnoea or OSA for a single night. A post hoc analysis revealed a subgroup of “responders” in whom ventricular ectopy decreased during sleep. However, only a few subjects had OSA and only two of the eight subjects with OSA had reductions in both AHI and VPBs. That study was therefore inconclusive regarding the effects of CPAP on ventricular ectopy in CHF patients with OSA.
Approximately one third of patients with HF die suddenly and, in 50% of these cases, this is due to ventricular tachyarrhythmias.8 Gami et al33 recently reported a significantly higher peak sudden death rate in patients with OSA during sleep time (12 midnight to 06.00 hours) than in patients without OSA in whom the peak sudden death rate occurred later in the morning after sleep. This suggests that OSA was playing a role in triggering sudden death during sleep, most likely through ventricular arrhythmias. Such arrhythmias could therefore pose a long term hazard in untreated OSA patients with HF. We included subjects with more than 10 VPBs/h because it has been shown that VPBs at this rate constitute a risk for malignant ventricular arrhythmias and both sudden death and overall mortality.9,22,32 Treatment of OSA, especially in patients with VPBs during sleep, therefore has the potential to prevent sudden death at night partly through suppression of such arrhythmias. Our finding that alleviation of OSA by CPAP suppressed VPBs supports this possibility.
The strengths of our study were that it was randomised and the results were clearcut and very consistent among individuals: the frequency of VPBs fell in all patients randomised to CPAP but did not change in the control group. Despite randomisation, the AHI and frequency of arousals were greater and minimum Sao2 lower in the control group than in the CPAP group. These imbalances probably arose because of the small number of individuals involved with more than 10 VPBs/h. However, there was no correlation between the frequency of VPBs and either the AHI or minimum Sao2. Moreover, the frequency of VPBs decreased in all subjects randomised to CPAP irrespective of their baseline AHI and frequency of VPBs. Thus, baseline AHI, minimum Sao2, and frequency of VPBs did not influence the response to CPAP. This, together with the fact that two way analysis of variance takes into account baseline differences in assessing time-treatment interactions, indicates that between group differences in these baseline variables are not likely to have influenced the primary outcome of our study. Because we observed only very few episodes of couplets or non-sustained ventricular tachycardia, we could not ascertain whether treating OSA reduced the frequency of such events.
Pharmacological interventions that have shown almost complete suppression of VPBs (>90%) have been found to have either an adverse or no effect on mortality.9,33 Several specific anti-arrhythmic agents are negative inotropes and may lead to the progression of HF.9 However, other drugs such as β blockers, which reduce mortality, cause a reduction in VPBs of only approximately 70%,10,11,34 similar to the 58% reduction observed in response to CPAP in our study. Beta blockers act by blocking cardiac β1 receptor stimulation, thereby reducing cardiac energy expenditure, heart rate and ventricular ectopy, increasing LVEF, and reducing both overall and sudden death rates. Although not proven, part of this reduction in mortality is probably due to a fall in ventricular ectopy.10,11 Similar to β blockers, CPAP acts not by directly suppressing arrhythmogenicity but by inhibiting autonomic, chemical, and haemodynamic stimuli shown to incite ventricular ectopy in heart failure. This non-pharmacological approach might therefore spare patients the adverse effects of antiarrhythmic drug therapy. The CPAP induced reduction in nocturnal sympathetic activity and VPBs and the improvement in LVEF observed in our study are analogous to the effects of β blockers and suggest the potential for CPAP to improve cardiovascular outcomes of patients with HF and OSA.
Our study was restricted to the effect of CPAP on VPBs during sleep. As OSA is limited to sleep, overnight monitoring was performed to test the effects of alleviation of OSA on VPBs specifically during this period. A longer monitoring period would allow us to determine whether this suppression of VPBs extends into the daytime.
In summary, we found that in patients with both HF and OSA, the application of therapeutic CPAP for 1 month causes a highly significant reduction in VPBs during sleep. These data imply that OSA can provoke VPBs during sleep, and that this stimulus is amenable to specific non-pharmacological therapy. Although no data are available on the prognostic significance of OSA in HF, it is possible that OSA induced ventricular ectopy could contribute to increased cardiovascular events in such patients with HF. Our data also suggest the possibility that reductions in VPBs and other ventricular arrhythmias through treatment of OSA could improve the prognosis of HF patients. However, larger scale, longer term studies will be required to test this hypothesis.
REFERENCES
Footnotes
-
Published Online First 30 June 2005
-
This study was supported by operating grant MOP-11607 from the Canadian Institutes of Health Research (CIHR). CMR is supported by research fellowships from Respironics Inc and the Toronto Rehabilitation Institute, KU by a research fellowship from Respironics Inc, JSF by the Canada Research Chair in Integrative Cardiovascular Biology and a Career Investigator Award from the Heart and Stroke Foundation of Ontario, and TDB by a Senior Scientist Award from the CIHR.
-
Competing interests: CMR and KU received research fellowship funding from Respironics Inc, and JSF and TDB have received research funding for an unrelated clinical trial involving heart failure patients with central sleep apnoea from Respironics Inc, ResMed Inc, and Tyco Healthcare Inc.