Article Text

Abstract
Pulmonary capillary haemangiomatosis is a rare disorder characterised by multiple angiomatous lesions composed of proliferating capillary vessels in the lung parenchyma that usually progress rapidly to establish fatal pulmonary hypertension. The 29 year old man presented here, however, has been stable for 3.5 years since the diagnosis without symptoms of pulmonary hypertension. High resolution computed tomographic findings of the pulmonary lesions seemed specific to the disease.
- pulmonary capillary haemangiomatosis
- high resolution computed tomography
- pulmonary hypertension
Statistics from Altmetric.com
Pulmonary capillary haemangiomatosis (PCH) has been recognised as a rare cause of pulmonary hypertension.1Patients with the disease typically present with pulmonary hypertension and abnormal chest radiographs.2 The condition usually progresses rapidly until severe pulmonary hypertension and resulting right cardiac failure develop.2 We report a case who presented with the disease without evident pulmonary hypertension, in whom no progression of symptoms or imaging findings of the pulmonary lesions has been observed over a long period.
Case report
A 29 year old man presented with haemoptysis and was admitted to hospital. He had been in excellent health until about 2 weeks earlier when he developed a sore throat and low grade fever (37.2°C) accompanied by occasional small amounts of haemoptysis. About 10 days before admission the symptoms suggesting an upper respiratory tract infection had disappeared but he continued to cough up small amounts of bright red blood without evident sputum. On admission the physical examination was normal and blood counts, erythrocyte sedimentation rate, C reactive protein, liver and renal function tests including urinalysis were also normal. Pao 2 was 11.6 kPa and Paco 2 was 5.6 kPa. Pulmonary function tests were normal except for a slight decrease in carbon monoxide transfer factor (Tlco) to 7.6 mmol/min/kPa (71% of predicted value). Extensive serological examinations failed to find any indications of angiitis, collagen vascular autoimmune disease, or HIV infection. The electrocardiogram and echocardiogram were both normal, without any findings suggestive of pulmonary hypertension such as tricuspid regurgitation. Chest radiography revealed patchy ground glass opacities scattered mainly in the bilateral lower lung field. A computed tomographic (CT) scan of the chest demonstrated 55 lesions in the right lung field and 33 in the left lung field (fig 1A). High resolution CT (HRCT) images showed that each lesion consisted of three layers, with focal nodular opacity at its centre surrounded by ground glass opacity and thickened interlobular septae at its margin (fig 1B). Transbronchial lung biopsy specimens revealed non-specific pulmonary haemosiderosis. The bronchoalveolar lavage fluid was macroscopically normal with normal cell counts.

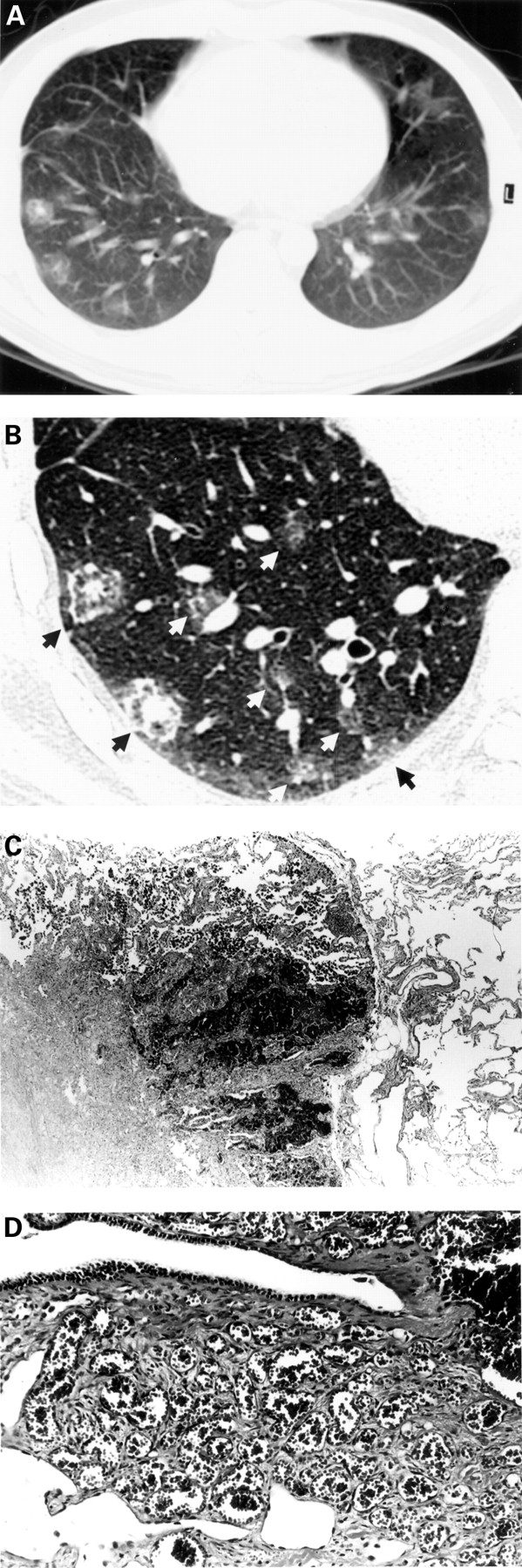{kind=link}
(A) Conventional CT scan (both collimation and interval 10 mm) of the patient revealed multiple pulmonary lesions and HRCT scan (both collimation and interval 1 mm) disclosed detailed structure of the lesions. (B) A section through the HRCT scan showed eight pulmonary lesions (arrows). (C) Microscopic study of a lesion showed fibrosis at its centre surrounded by intra-alveolar haemorrhage (or congestion) and thickened interlobular septae (haematoxylin/eosin, ×12.5). The structure of each lesion seemed to reflect the nodular opacity at its centre, surrounding ground glass opacity, and its thickened margin, respectively, on the HRCT image. (D) High magnification of the lesion revealed proliferation and infiltration of capillaries in the interstitium (haematoxylin/eosin, ×50) and confirmed the diagnosis of pulmonary capillary haemangiomatosis.
Open lung biopsy of the right thorax was performed. Inspection of the visceral pleura disclosed numerous dark red subpleural lesions mainly in the middle and lower lobes. Microscopic examination of the biopsy specimens revealed extensive intra-alveolar haemorrhage, both recent and old. The alveolar septae in the areas of old haemorrhage showed mild fibrous thickening accompanied by chronic inflammatory cells. Fibrotic scarring was observed at the centre of the lesions. In the adjacent lung parenchyma there was evidence of proliferation of capillaries within the alveolar septae and inside the pulmonary artery wall. These lesions were sharply demarcated by interlobular septae (fig1C, D). These findings established the diagnosis of PCH.
The patient refused further invasive examinations including cardiac catheterisation, angiography, and a new treatment approach with interferon α. He has had no symptoms except for an occasional small amount of haemoptysis during the 3.5 years since the first admission. He now swims 600 m several times a week. A recent 6 minute walking distance was 620 m and there was no decrease in Spo 2 during this exertion. The most recent Pao 2 was 11.6 kPa and Paco 2 was 5.2 kPa. Conventional and HRCT scans performed 40 months after initial admission showed no increase in the number of lesions, which remained unaltered in size.
Discussion
This case of PCH appears to be unusual in that the condition has remained stable for a long period without symptomatic pulmonary hypertension. Although the lack of clinical signs suggestive of pulmonary hypertension and the patient's high exercise performance preclude the presence of severe pulmonary hypertension, the existence of minor pulmonary hypertension cannot be ruled out because of a lack of cardiac catheterisation data. When considering the nature of PCH, the patient may have—and, indeed, probably does have—increased pulmonary pressure prior to the development of clinical signs of pulmonary hypertension.
PCH was initially described in 19783 and Lippertet al 4 found only 20 reported cases including their own in approximately 20 years. Al-Fawazet al 2 reviewed 19 cases which appeared in the literature. Briefly, PCH is characterised by the proliferation and infiltration of small vessels, mostly capillary sized, within the peribronchial, perivascular, septal or pleural connective tissues. Involvement of the airways probably leads to haemoptysis. Without effective treatment the condition usually progresses rapidly, causing death from pulmonary hypertension and/or bleeding. Of the 19 cases, four underwent pulmonary (with or without heart) transplantation, one had pneumonectomy, and one was treated with interferon α-2a. These treatments reportedly resulted in improvements in their conditions. Furthermore, one of the cases did, in fact, have a short period of observation.2 The other 12 cases died within a total clinical course ranging from several months to several years, and all showed progression of dyspnoea, exercise intolerance, cardiac failure, and/or haemoptysis during this period.
In contrast to the active and progressive nature of the disease in the above cases, our case has been followed up for 3.5 years since the diagnosis of PCH during which there has been no clinical deterioration or aggravation of the imaging findings. To our knowledge, this is the first report of a case with stable disease without any treatment for such a long period. Although two cases reported by Whiteet al 1 and Vevainaet al 5 also had long retrospective clinical courses, before the diagnosis of PCH detailed correlation between PCH and the preceding symptoms was not established; the former had recurrent chest infection for 12 years and the latter had digital clubbing for 4 years. Our case possibly represents an early phase of such cases.
Some reports have discussed the possibility that radiological features may be unreliable in separating PCH from other diseases.5 ,6 In our case, however, conventional and HRCT findings differentiated the condition from commonly seen diseases, and this fact urged us to proceed to the open lung biopsy. Three previously reported cases who underwent HRCT scanning had findings similar to ours.7 ,8 Two other cases studied solely by conventional CT scanning2 ,4 also reported findings that were consistent with ours. As in our case, the HRCT findings of the patient reported by Dufour et al 7 seem to be specific to PCH, demonstrating the usefulness of HRCT scanning for diagnosis of this disease. Other previously reported CT findings of the disease include increased soft tissue density of the mediastinum1 ,2 and mediastinal and hilar lymph adenopathies,7 but such findings were absent in our case.
Possible explanations for the stable condition of our case over a long period are either that it was a chance discovery of an early phase of the disease by HRCT scanning or that PCH has separate subtypes with differing speeds of progression. Wider use of HRCT scanning and more extensive understanding of PCH would resolve this issue in the future.
Acknowledgments
The authors thank Professor Rubin M Tuder (Department of Pathology-Surgical Pathology Laboratory, University of Colorado Health Sciences Center) and Professor Takesaburo Ogata (Center for Medical Sciences, Ibaraki Prefectural University of Health Sciences) for their pathological review of the case. This work was supported by funding from the Ministry of Education of Japan.