Article Text

Statistics from Altmetric.com
- hypoxia
- oxygen sensing
- hypoxia inducible factor-1α (HIF-1α)
- prolyl hydroxylase domain containing enzymes (PHDs)
- ARNT, aryl hydrocarbon receptor nuclear translocator
- FIH, factor inhibiting HIF
- HIF, hypoxia inducible factor
- HRE, hypoxic response element
- PHD, prolyl hydroxylase domain containing enzyme
The oxygen sensing pathway offers a new set of therapeutic targets for conditions ranging from inflammatory lung disease to pulmonary hypertension
The ability of cells to detect and respond to a fall in oxygen tension is of fundamental importance for maintaining oxidative metabolism and tissue homeostasis. One of the challenges facing scientists working in this area has been that any proposed mechanism for oxygen sensing has to accommodate the very different tolerances of certain tissues to hypoxia and the extreme variation in the cellular responses observed. Hence, while skeletal muscle cells can recover function after 30 minutes of anoxia, the brain suffers irreparable damage after only 4–6 minutes of ischaemia.1 Moreover, while carotid body cells respond to changes in oxygen tension that barely register in non-chemosensory tissues (and do so within seconds),2 upregulation of erythropoietin synthesis in the interstitial peritubular cells is transcriptionally regulated and requires far more protracted periods of hypoxaemia.3 Despite such variances in oxygen sensitivity and response time, all cells appear capable of responding to hypoxia and the essential components of a universal oxygen sensing mechanism have at last begun to emerge. Moreover, from studies conducted in stroke and heart disease, it is apparent that therapeutic targeting of this novel pathway is set to transform our approach to pathology previously deemed intractable.
RESEARCH STUDIES
Initial clues into how cells respond to low oxygen came from studies undertaken in the early 1990s examining the hypoxic response element (HRE) of the erythropoietin (Epo) gene. This led to the identification of a transcriptional activator called hypoxia inducible factor (HIF).4 HIF is a heterodimer composed of HIF-1β (or aryl hydrocarbon receptor nuclear translocator, ARNT), which is constitutively expressed, and HIF-1α whose expression and transcriptional activity are tightly regulated by the ambient oxygen concentration.5 Once formed, this protein complex migrates to the cell nucleus and, together with the co-activator CBP/p300, binds to the HREs present on the promoter region of genes involved in regulating metabolic supply and demand.6 Examples of HIF regulated genes include those involved in regulating vascular tone (for example, iNOS and adrenomedullin), angiogenesis (for example, vascular endothelial growth factor, VEGF), cell metabolism (for example, lactate dehydrogenase A, the glucose transporter GLUT-1), and haemoglobin biosynthesis (for example, erythropoietin).7 These findings, together with the ubiquitous nature of HIF and the demonstration that HIF deficient animals show major defects in many core physiological responses to oxygen, have resulted in HIF being regarded as one of the master regulators of the cellular response to hypoxia.8
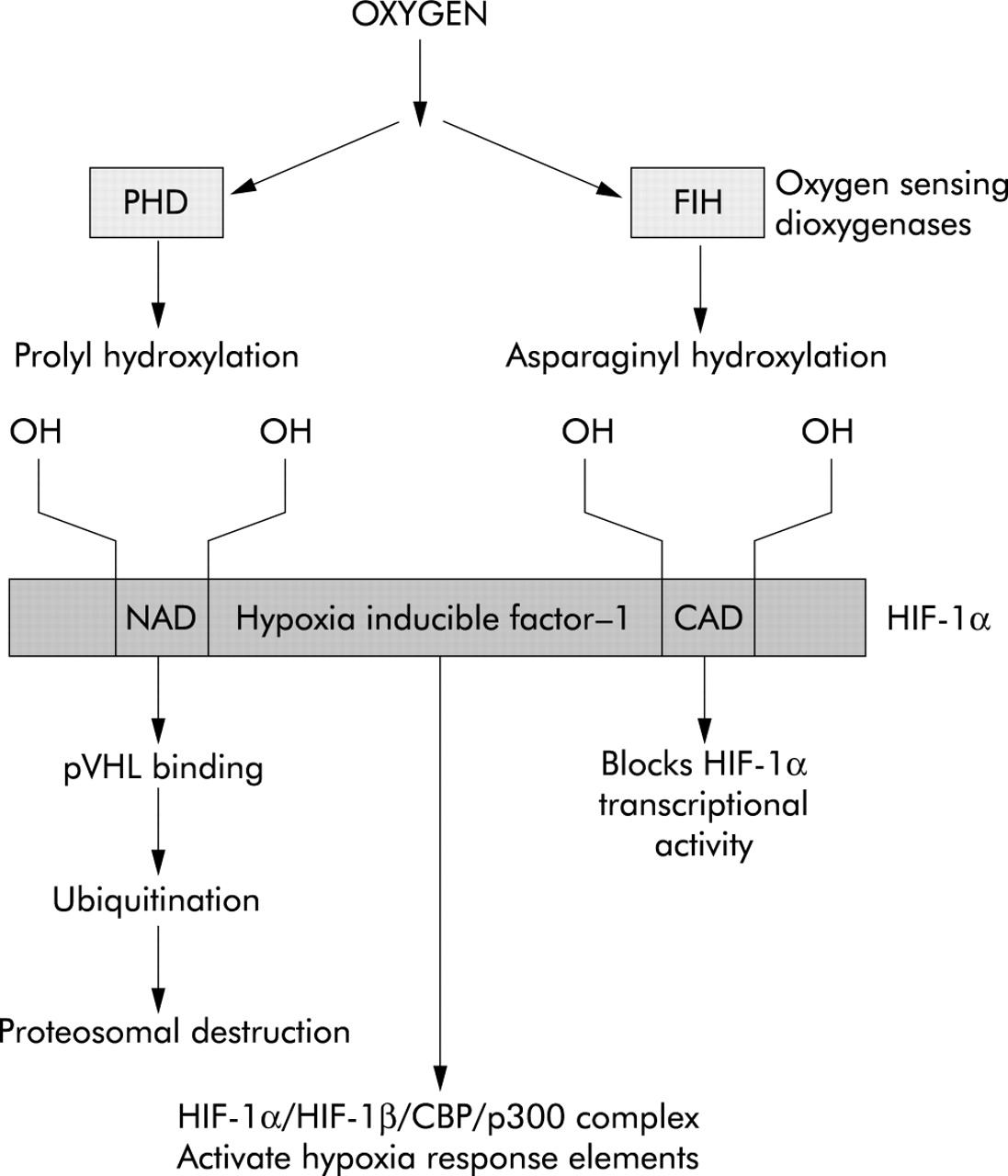
The next step in this quest was to define the mechanism responsible for hypoxic induction of HIF-1α. Through a combined structural and genetic approach, it has now been possible to show that HIF-1α activity is regulated by enzymatic hydroxylation at specific prolyl and asparaginyl residues by a novel 2-oxoglutarate dependent class of dioxygenases.9 Critically, these enzymes have been shown to display an absolute requirement for oxygen in addition to iron (Fe2+) and ascorbate. These oxygen sensitive enzymes inhibit HIF activity in a complementary manner since the prolyl hydroxylase domain containing enzymes (PHDs) result in an interaction between HIF-1α and the von Hippel-Lindau protein which targets HIF-1α for proteosomal destruction,10 and factor inhibiting HIF (FIH) causes asparaginyl hydroxylation and blocks HIF association with CBP/p300.11,12 Hence, under normoxic conditions, HIF-1α levels remain low and this prevents the transcription of genes containing HRE promoters (fig 1).

{kind=link}
Oxygen dependent inhibition of HIF-1α. Under normoxic conditions the oxygen requiring PHD and FIH dioxygenases lead to prolyl and asparaginyl hydroxylation of HIF-1α which results in proteosomal destruction and inhibition of transcriptional activity. PHD, prolyl hydroxylase domain containing enzyme; FIH, factor inhibiting HIF; NAD, amine activation domain; CAD, carboxyl activation domain; pVHL, von Hippel-Lindau protein.
CLINICAL APPLICATIONS
How does this inform our understanding of the pathophysiology of lung disease and can it provide the basis of novel therapies? Mice homozygous for a null mutation in the HIF-1α13 or HIF-1β genes14 die at mid gestation with vascular defects primarily involving the embryonic and extraembryonic circulation, respectively. In contrast, HIF-1α+/− mice develop normally and are indistinguishable from wild type littermates. However, when exposed to 10% oxygen for up to 6 weeks, the HIF-1α+/− mice demonstrate reduced susceptibility to pulmonary hypertension, polycythemia, and right ventricular hypertrophy relative to their wild type littermates.15 Morphometric analysis showed that the chronically hypoxic HIF-1α+/− mice have fewer completely muscularised pulmonary arterioles and the degree of muscularisation in such vessels is reduced compared with HIF-1α wild type mice. Thus, HIF-1 appears to play a major role in mediating pulmonary vascular remodelling in chronic hypoxia, and therapeutic manoeuvres that inhibit HIF-1 activity in the lung may slow the progression of hypoxia induced pulmonary hypertension.
Likewise, in many human cancers including primary lung tumours, increased levels of HIF-1α expression are associated with resistance to tissue hypoxia and enhanced tumour angiogenesis.16 This abnormality correlates with a reduced sensitivity to radiation and chemotherapy and disease progression.17 Moreover, in vitro work has shown that depleting tumour cells of HIF-1 renders them more radioresponsive and thus provides an opportunity to enhance the therapeutic potential of conventional treatment.18 In addition, by targeting bioreductive pro-drugs to hypoxic tumour sites, the potential for normal tissue damage can be reduced.
One of the real surprises in this area of research, however, has been the recent demonstration of the importance of oxygen sensing pathways in the function of inflammatory cells. Hence, in contrast to the effects of hypoxia observed in many other cell types, oxygen deprivation causes a profound but reversible delay in the rate of constitutive apoptosis in human neutrophils.19 Moreover, removing the capacity of neutrophils to respond to hypoxia using a myeloid targeted HIF-1α knock-out results in a substantial fall in the glycolytic capacity of these cells and, as a consequence, a profound impairment of myeloid cell adhesion migration and bacterial killing.20 As persistent accumulation of primed and activated neutrophils is a cardinal feature of a number of lung diseases, these data predict that inhibition of the HIF-1α pathway may promote the resolution of granulocyte inflammation.
One final example of the importance of this pathway relates to the potential to prevent ischaemia-reperfusion injury in the transplanted lung. Hence, in the heart, ischaemic pre-conditioning, which reduces dramatically the degree of myocardial damage observed following subsequent coronary artery occlusion, has been shown to relate to HIF dependent upregulation of erythropoietin.21 Indeed, this protein has the capacity to prevent myocardial cell apoptosis, and erythropoietin infusion is effective in reducing infarct size and improving outcome in stroke.22 Application of these principles to lung protection is bound to follow.
Despite such striking advances in our knowledge of how cells sense oxygen, much remains to be defined. In particular, it is likely that a quite separate pathway underlies the ability of acute hypoxia to induce membrane depolarisation and l-type calcium channel activity in excitable cells. These events trigger the acute contractile response of pulmonary artery resistance vessels to hypoxia and the firing of carotid body glomus cells. The current contenders in this area are the mitochondrial NAD(P)H oxidases, which are postulated to produce a diffusible redox mediator in response to hypoxia.
CONCLUSIONS
These studies show that, while cells display widely differing tolerances and responses to hypoxia, oxygen sensing by dioxygenases and their consequent effects on HIF dependent transcriptional events appear to be a property that is shared by most, if not all, cells. This pathway now offers a new set of therapeutic targets for an array of lung diseases ranging from inflammatory lung disease through to pulmonary hypertension.
Acknowledgments
The work in the authors’ laboratories is sup-ported by the Wellcome Trust, British Heart Foundation, National Asthma Campaign, MRC, and British Lung Foundation. SRW holds an MRC Clinical Research Training Fellowship and KKS holds a BHF Clinical Fellowship.
The oxygen sensing pathway offers a new set of therapeutic targets for conditions ranging from inflammatory lung disease to pulmonary hypertension