Article Text

Abstract
Background Birt-Hogg-Dubé syndrome (BHD) is a clinical syndrome manifesting with cystic lung disease and pneumothorax. Features of BHD result from the loss-of-function mutations of the folliculin (FLCN) gene. Chronic obstructive pulmonary disease (COPD), characterised by an irreversible airflow limitation, is primarily caused by cigarette smoking.
Objective Given that COPD often shares structural features with BHD, we investigated the link between COPD, cigarette smoke (CS) exposure and FLCN expression.
Methods We measured the expression of FLCN in human COPD lungs and CS-exposed mouse lungs, as well as in CS extract (CSE)-exposed immortalised human airway epithelial cells by immunoblotting.
Results We found that the lung FLCN protein levels in smokers with COPD and CS exposure mice exhibit a marked decrease compared with smokers without COPD and room air exposure mice, respectively. We confirmed CS induced degradation of FLCN in immortalised human bronchial epithelial Beas-2B cells via ubiquitin proteasome system. Further, siRNA targeting FLCN enhanced CSE-induced cytotoxicity. By contrast, FLCN overexpression protected cells from CSE-induced cytotoxicity. We found that FBXO23, the ubiquitin E3 ligase subunit, specifically binds to and targets FLCN for degradation. Inhibition of ATM (ataxia‐telangiectasia mutated) attenuated CSE induced FLCN degradation, suggesting a role of ATM in FLCN proteolysis. We further confirmed that the mutant of major FLCN phosphorylation site serine 62A is resistant to CSE-induced degradation and cytotoxicity.
Conclusions Our study demonstrates that CS exposure is a secondary cause of FLCN deficiency due to the enhanced proteolysis, which promoted airway epithelial cell death.
- airway epithelium
- emphysema
- rare lung diseases
- tobacco and the lung
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
Birt-Hogg-Dubé syndrome due to the genetic deficiency of folliculin (FLCN) is manifested by multiple lung cysts and spontaneous pneumothorax, which can be seen among some smokers. However, the effects of smoking on FLCN expression have not been investigated.
What is the bottom line?
Cigarette smoking depletes FLCN in the lung of smokers with chronic obstructive pulmonary disease (COPD) and in the lung of mice. Cigarette smoke exposure destabilises FLCN through phosphorylation at serine 62 via the ubiquitin-proteasome mechanism.
Why read on?
Cigarette smoking is a secondary cause of FLCN deficiency. Future studies will be needed to investigate the potential causal role of smoking-induced FLCN loss in the development of COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive destructive lung disease characterised by airway inflammation, remodelling and emphysema, leading to obstructive ventilatory defects.1 Cigarette smoking is a major risk factor for COPD. COPD is a widespread and increasing global health problem and affects greater than 10% of the world population over the age of 40 years, resulting in a substantial economic and social burden.2 3 Unfortunately, there is still no effective treatment that significantly reduces disease progression or mortality.
Birt-Hogg-Dubé syndrome (BHD), a rare autosomal dominant condition due to the germline loss-of-function mutations of folliculin (FLCN) gene, is characterised by benign tumours of the skin, lung cysts, spontaneous pneumothorax and increased risk of renal carcinoma.4 5 Moreover, smokers with BHD are complicated by more prominent cystic lung disease relative to non-smokers with BHD.6 However, the underlying mechanism of lung cyst formation in BHD is still unclear. FLCN harbours structural features of small GTPase regulators and plays an important role in diverse cellular processes, including mammalian target of rapamycin (mTOR) pathway and cell–cell adhesion.7 8 In addition to the activation of the mTOR pathway,9 deficiency of FLCN results in defection of cell–cell interaction and impairment of cellular repair mechanisms in lung damage.7 10
Several studies have focused on elucidating the specific pathway(s) that may link FLCN to lung cyst formation. Alveolar epithelium-specific FLCN-knockout mice exhibit lung morphological changes that resemble emphysema (ie, alveolar destruction or enlargement)10 11 which indicate the implication of FLCN in maintaining the integrity of alveolar structure. FLCN deletion in mouse lung epithelium leads to cell apoptosis and an impairment of both epithelial barrier and overall lung function.11 Loss of FLCN may contribute to dysregulation of lung tissue structure and homeostasis that ultimately results in lung cyst formation, mimicking bullous emphysema, often seen in smoking-induced COPD.12 However, it remains unclear whether COPD and smoking affect FLCN function.
Cigarette smoke (CS) exposure often alters protein stability.13 14 Proteolysis is regulated by the two major degradation pathways in eukaryotic cells termed the ubiquitin proteasome system (UPS) and the lysosomal pathway.15 The ubiquitin cascade forms a ubiquitin-substrate complex that is tagged for degradation which requires the action of at least three ubiquitin enzymes including the ubiquitin activator (E1), the ubiquitin conjugating enzyme (E2) and ubiquitin ligase (E3). Among these three different ubiquitination enzymes, the E3 ubiquitin ligases recognise and recruit specific substrates.16 The Skp1 (S phase kinase-associated protein 1)-Cul1 (cullin 1)-F-box protein (SCF) complexes is one of the best characterised E3 ligase family. The variable F-box proteins determine the substrate specificity by recruitment of substrates for ubiquitination. By targeting diverse substrates, F-box proteins exert control over stability of proteins and regulate the mechanisms for a wide range of cellular processes.17 18
In this study, we investigated a potential link between BHD and smoking-induced COPD. Our findings show that cigarette smoking downregulates FLCN in the lung, which may contribute to lung epithelial cell death and possibly the development of emphysema.
Materials and methods
Cell lines and reagents
Human primary alveolar epithelial cells (pHAECs) were purchased form Cell Biologics (Cat#: H6053) and maintained according to the manufacturer’s instruction. Primary human bronchial epithelial cells (pHBECs) were provided by the tissue culture core of the University of Pittsburgh. Bronchial epithelial Beas-2B cells were cultured in hydrocortisone, insulin, transferrin, oestrogen and selenium medium (500 mL of Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 [DMEM/F12], 2.5 mg of insulin, 2.5 mg of transferrin, 2.5 mg of sodium selenite, 2.5 mg of transferrin, 10 µM hydrocortisone, 10 µM β-oestradiol, 10 mM Hepes and 2 mM L-glutamine) containing 10% fetal bovine serum and maintained in a 37°C incubator in the presence of 5% CO2. Radioimmunoprecipitation assay (RIPA) Lysis and Extraction Buffer (Cat#: 89901), ataxia‐telangiectasia mutated (ATM) siRNA (Cat#: 1299001), V5 antibody (Cat#: PA1-993) and Escherichia coli Top10 competent cells (Cat#: C404010) were purchased from Invitrogen (St Louis, Missouri). FLCN (Cat#: 3697S) and haemagglutinin (HA) tag (Cat#: 3724S) antibodies were purchased from Signalling (Danvers, Massachusetts). Ubiquitin (Cat#: sc-166553) antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, California). Cycloheximide (Cat#: ALX-380–269 G001) and Ubiquitin aldehyde (Cat#: BML-UW8450-0050) were obtained from Enzo Life Sciences (Farmingdale, New York). The β-Actin antibody (Cat#: A3853) and the protease inhibitors leupeptin (Cat#: CAS 103476-89-7) and MG132 (Cat#: 133407-82-6) were obtained from Sigma (Carlsbad, California). Goat Anti-Rabbit IgG (Cat#: 1706515) and Goat Anti-Mouse IgG (Cat#: 1706516) were obtained from Bio-Rad Laboratories (Hercules, California). High-Capacity RNA-cDNA Kit (Cat#: 4387406), Alexa Fluor 488 Donkey anti-Rabbit Ig (Cat#: A-21206) and SYBR Select Master Mix (Cat#: 4472942) were purchased from Thermo Fisher Scientific (Waltham, Massachusetts). FLCN siRNA (D-009998-03-002) was obtained from Dhmarmacon (Lafayette, Colorado). QuikChange II XL site-directed mutagenesis kits (Cat#: 200522) were obtained from Agilent Technologies (Santa Clara, California). Nu7441(Cat#: 503468-95-9), VE-821 (Cat#: 1232410-49-9) and Ku-55933 (Cat#: 587871-26-9) were obtained from Cayman Chemical (Ann Arbor, Michigan). All other reagents were of the highest grade commercially available.
Human lung tissue samples
The lung tissue research consortium and the tissue core of the University of Pittsburgh provided frozen lung parenchymal tissue from smokers both with very severe COPD (N=6) and without known lung diseases (N=5), respectively (table 1).
The demographic data for the human subjects evaluated for lung FLCN protein
Animals
The animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh School of Medicine. C57Bl/6 mice obtained from Charles River were exposed to 6 months of either CS (N=4) or room air (RA) (N=4) starting at 8–10 weeks of age, as previously described. The CS group was exposed to four cigarettes/day, 5 days/week. The RA group was exposed to room air alone.
Preparation of CS extract
Research cigarettes (3R4F) were purchased from the University of Kentucky. CS extract (CSE) was prepared as we previously described.13 14 19
Plasmid and siRNA transfection
Beas-2B cells were transfected with plasmids or siRNA using electroporation executed with a nuclear transfection apparatus (Amaxa Biosystems, Gaithersburg, Maryland). The nuclear transfector was preset to T-013, a programme for Beas-2B cells. About 2 µg of plasmid or 40 pm of siRNA was electroporated into 1 million cells and cultured in a six-well plate for further analysis.
Immunoblot analysis and immunoprecipitation
Lung tissue was cut into appropriately sized pieces, washed in cold phosphate buffered saline (PBS) three times, mixed with two volumes of RIPA lysis buffer supplemented with protease and phosphatase inhibitors and homogenised with steel using a Bullet Blender on speed 10 and time 4. Tissue lysates were centrifuged at 13 000×g for 10 min. Cells were harvested, lysed in ice–cold RIPA, sonicated and centrifuged. Lysates were cleared by centrifugation and analysed by gel electrophoresis. Protein concentration was determined using a Bradford protein assay (Bio‐Rad, Hercules, California, USA). Tissue or cell lysates were resolved by SDS-PAGE and transferred to the nitrocellulose membrane. The membranes were blocked with non-fat milk and probed with indicated primary antibodies and then developed with enhanced chemiluminescence system.
For immunoprecipitation, about 1 mg cell lysates (in PBS with 0.5% Tween 20 plus protease inhibitors) were incubated with a specific primary antibody overnight at 4°C. The mixture was added to 40 µl of protein A/G agarose for 2 hours at room temperature. The precipitated complex was washed with cold PBST (PBS+0.1% Tween 20) three times for 5 min each and analysed by immunoblotting.
Quantitative PCR
Quantitative PCR (qPCR) experiments were performed using the standard SYBRTM Select Master Mix Kit protocol and run in a CFX96TM Real-Time System (Bio-Rad, USA). The primer pairs were commercially synthesised (Sigma, see table 2). The reactions were incubated in a 96-well plate at 95°C for 2 min, followed by 40 cycles of 95°C for 15 s, 58°C for 15 s, and 72°C for 1 min. All reactions were run in triplicate. The target messenger RNA (mRNA) expression is normalised on the expression of the reference gene and given as 2−ΔCt (ΔCt=Cttarget−Ctreference), relative changes of treated group compared with control are 2−ΔΔCt values (ΔΔCt=ΔCttreated−ΔCtcontrol).
RT-PCR primers
Site-directed mutagenesis
QuikChange II XL site-directed mutagenesis kit was used to introduce mutagenesis according to the manufactural instructions. Briefly, mutant strands were synthesised by PCR using the pcDNA-FLCN-HA plasmid as the template and one pair primer containing the desired mutation of FLCN-S62A-F-GATGCGTGCGCACGCCCCCGCAGAGGGGG and FLCN-S62A-R -CCCCCTCTGCGGGGGCGTGCGCACGCATC. The PCR product was treated by Dpn I to digest the parental DNA template and transformed into XL1-Blue supercompetent cells. The accuracy of site-directed mutagenesis was confirmed by Sanger sequencing provided by GENEWIZ.
Statistical analysis
Results are expressed as a median with IQR or mean±SEM. The statistical differences between groups were determined by the Mann-Whitney U test, Student’s t-test or one-way analysis of variance with Bonferroni’s correction. A probability value of less than 0.05 was accepted as statistically significant.
Results
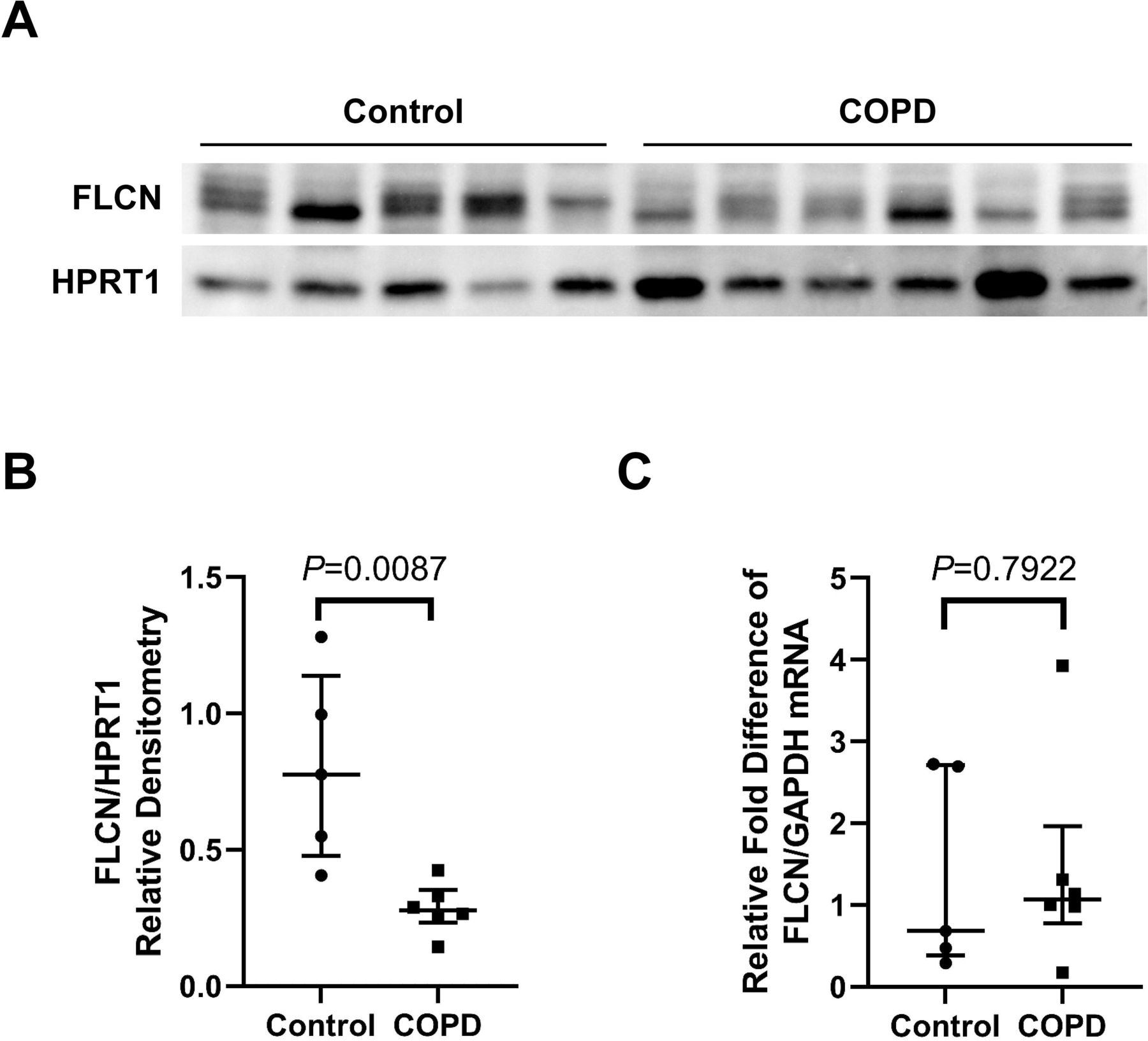
Smokers with COPD exhibit decreased expression of pulmonary FLCN protein
To determine the steady-state levels of FLCN mRNA and protein in the lung of smokers with COPD, whole lung parenchymal tissue from smokers with very severe COPD (n=6) were evaluated and compared with smokers without known lung diseases (n=5). FLCN protein was markedly decreased in COPD lungs relative to control smokers’ lungs (figure 1A and B, all the original raw blots are available in online supplemental file 1). By contrast, qPCR data showed that mRNA levels of FLCN in the lungs of individuals with COPD were not significantly different (figure 1C) compared with lungs of smokers without COPD. We also measured FLCN protein levels in the lungs of non-smoking and actively smoking subjects without known lung disease (n=5/group). FLCN protein levels in the lungs of non-smoking is no significant change compared with active smokers (online supplemental figure 1A and B). These data suggest that the level of FLCN protein expression in lungs with COPD is decreased by a post-transcriptional mechanism.
Supplemental material
Supplemental material

Smokers with COPD exhibit decreased pulmonary FLCN (A): The stage of COPD was determined by the Global Initiative for Obstructive Lung Disease (GOLD) criteria. Stage 3, severe and stage 4, very severe. Smokers with normal lung function serve as control. Whole lung parenchyma lysates were prepared from a total of 11 smokers with various gold stages of COPD and analysed for FLCN by immunoblotting. (B): The densitometry data (FLCN/HPRT1) obtained from (A) are expressed as a median (lines) with IQR (IQR, whiskers) and analysed using Mann-Whitney test. (C): Total RNA was prepared from whole lung parenchymal tissues obtained from the same donors (control and stage 3/4) as in (A). Levels of FLCN mRNA were measured by qPCR. The relative fold difference compared with GAPDH (control) was expressed. Data are expressed as a median (lines) and IQR (whiskers). COPD, chronic obstructive pulmonary disease; FLCN, folliculin; mRNA, messenger RNA; qPCR, quantitative PCR.
CS depletes FLCN in mouse lungs
To determine the effect of CS exposure on FLCN expression, mice lungs were harvested and analysed to evaluate the alteration of FLCN. A marked decrease of FLCN was observed in mouse lungs exposed to CS for 6 months compared with RA control mice (figure 2A and B) by immunoblotting. We next measured the effect of CS on FLCN mRNA levels by qPCR. It showed that there is no significant difference of mouse lung FLCN mRNA between CS exposure and RA control (figure 2C). We also evaluated FLCN protein levels in mouse lungs exposed to CS for 6 weeks. There was no significant change compared with RA control (online supplemental figure 1C and D). These data also suggest that long-term CS exposure induces FLCN loss in mouse lungs via a post-transcription mechanism.
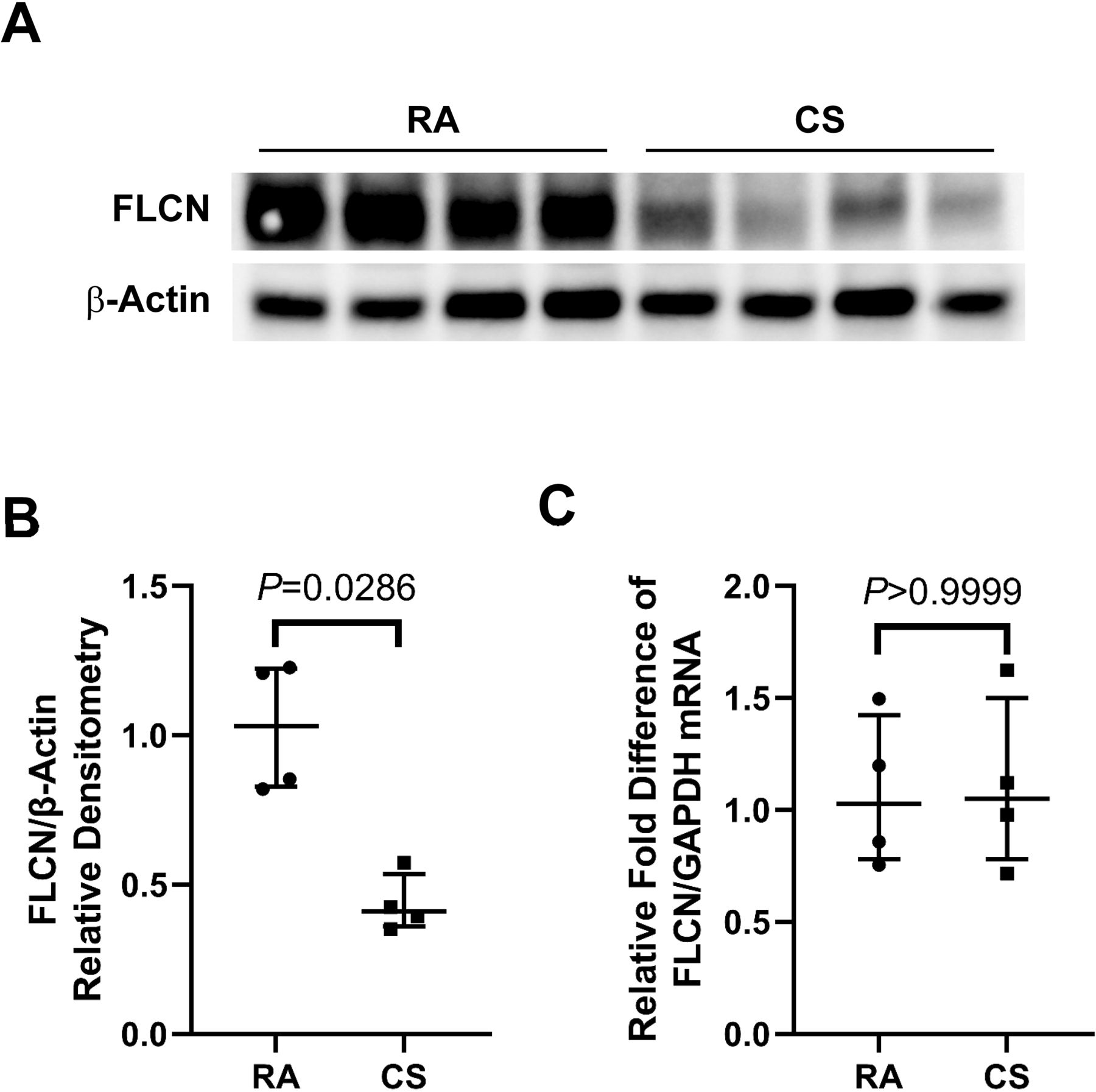
Pulmonary FLCN reduced in CS treated mice (A): Whole lung tissue were prepared from mice exposure to cigarette smoke (CS) or room air (RA) for 6 months. The samples were analysed by immunoblotting for FLCN. (B): The densitometry data (FLCN/β-Actin) obtained from (A) are expressed as a median (lines) with IQR (whiskers) and analysed using Mann-Whitney test. (C): Total RNA was prepared from whole lung tissues obtained from the same mice (RA and CS) as in (A). Levels of FLCN mRNA were measured by qPCR. The relative fold difference compared with GAPDH (control) was expressed. Data are expressed as a median (lines) and IQR (whiskers). FLCN, folliculin; mRNA, messenger RNA; qPCR, quantitative PCR.
FLCN protects against CS-induced cytotoxicity in BEAS-2B cells
To determine the effects of CS on FLCN protein in primary lung epithelial cells (LECs), both pHBECs and pHAECs were cultured with or without 2% CSE for 24 hours. CSE decreased FLCN protein levels in both primary cells (figure 3A). We further evaluated the effects of CS exposure on FLCN expression in Beas-2B cells. Immunoblotting showed that CSE decreased FLCN levels in Beas-2B cells in dose dependent manner (figure 3B). CSE reduced FLCN protein expression for up to 8 hours (figure 3C, D and E). Beas-2B cells were transfected with FLCN siRNA, and approximately 80% knockdown of FLCN was achieved (figure 3F). CSE-induced cytotoxicity was further augmented by FLCN protein depletion (figure 3G). We also generated HA-tagged FLCN plasmid (figure 3H and I). The ectopic FLCN overexpression (approximately 70% relative to the endogenous FLCN expression (empty vector control)) attenuated levels of CSE-induced cytotoxicity (figure 3J). These data suggest that CSE enhances FLCN protein degradation in Beas-2B cells in a dose-dependent and time-dependent manner. FLCN is a protect factor to CSE-induced cell toxicity.

FLCN is required to protect against cigarette smoke-induced cytotoxicity in BEAS-2B cells (A): Primary human alveolar epithelial cells (pHAECs) and primary human bronchial epithelial cells (pHBECs) were cultured with or without 2% CSE for 24 hours, and analysed by immunoblotting for FLCN or β-actin. (B): Beas-2B cells were exposed to different concentrations of fresh CSE (0, 1, 2, 4, 6%) for 8 hours, and analysed by immunoblotting for FLCN or β-actin. (C): BEAS-2B cells were treated with 2% CSE for up to 8 hours and analysed for FLCN or β-actin by immunoblotting. (D): The densitometry data (FLCN/β-Actin) obtained from (C) are expressed as mean±SEM. (E): Total RNA was prepared from cells as in (C). Levels of FLCN mRNA were measured by qPCR. The relative fold difference compared with GAPDH (control) was expressed. Data are expressed as mean±SEM. (F): BEAS-2B cells were transfected with siCrtl or siFLCN for 72 hours and cell lysates were immunoblotted for FLCN, or β-actin antibodies. (G): BEAS-2B cells were transfected with FLCN siRNA (siFLCN) or scrambled siRNA (siCtrl) for 48 hours and further cultured with or without 2% of CSE for 24 hours. MTT assay was performed for analysis of cell viability. (H): BEAS-2B cells were transfected with vector or FLCN-HA plasmid for 48 hours, and cell lysates were immunoblotted with FLCN or β-actin antibodies. Immunoblotting data are representative of three independent experiments. (I): BEAS-2B were transfected with FLCN-HA plasmid for 24 hours, and cells were fixed and stained with HA and Alexa Fluor 488 antibodies. (J): BEAS-2B cells were transfected with HA-tagged FLCN (FLCN) or empty vector (vector) for 24 hours and further cultured with or without 2% of CSE for 24 hours. Cell viability was measured by MTT assay. Data are expressed as mean±SEM for three independent experiments with triplicate samples. CSE, cigarette smoke extract; FLCN, folliculin; HA, haemagglutinin; mRNA, messenger RNA; qPCR, quantitative PCR.
Cigarette smoke decreases FLCN in human lung epithelial cells via the UPS
The stability of FLCN in Beas-2B cells were determined with the protein biosynthesis inhibitor cycloheximide (CHX) chase assay. The results showing that the FLCN is a labile protein with a half life of about 6 hours in Beas-2B cells. To test which pathway is involved in FLCN degradation, Beas-2B cells were treated with a proteasome inhibitor MG132 or a lysosomal inhibitor leupeptin. The accumulation of FLCN induced by only MG132 suggests that the ubiquitin proteasomal pathway mediates FLCN degradation for its disposal in Beas-2B cells (figure 4A and B). We separated the phosphorylated form and unphosphorylated form of FLCN in CHX-treated BEAS-2B cells using 7.5% gel. The phosphorylated FLCN decreased faster (T1/2: 4 hours) than unphosphorylated FLCN (T1/2: 6 hours) (online supplemental figure 2A and B). In addition, we found that FLCN protein levels decreased in a dose-dependent manner (figure 4C) by introducing the HA‐tagged ubiquitin construct, suggesting that ubiquitin directs FLCN degradation.

FLCN undergoes proteasomal degradation (A): BEAS-2B cells were treated with CHX (20 µg/mL) or leupeptin (20 µM) or MG132 (40 µM) for indicated times. Cell lysates were subjected to FLCN and β-actin immunoblotting. (B): The densitometry data (FLCN/β-Actin) obtained from (A) are expressed as mean±SEM. (C): BEAS-2B cells were transfected with indicated amounts of HA-tagged ubiquitin plasmid and the cell lysates were immunoblotted for FLCN, HA, or β-actin antibodies. Immunoblotting data are representative of three independent experiments. CHX, cycloheximide; FLCN, folliculin; HA, haemagglutinin.
FBXO23 targets FLCN for proteasomal degradation
To identify the E3 ligase to specifically recognise and mediate FLCN degradation, we screened multiple V5-tagged F-box protein overexpression plasmids and analysed FLCN protein stability. The individual V5 expressions of multiple F-box proteins were varied and only FBXO23, FBXO41 and FBXO42 were adequately expressed in the cells (figure 5A). Among the three F-box proteins, FBXO23 was associated with slightly lower expression levels of FLCN (figure 5B). Additionally, FBXO23 overexpression decreased FLCN levels in a dose-dependent manner compared with FBXO41 control (figure 5C and D). We chose FBXO41 as the negative control because the V5 expression is equivalent to the FBXO23 vector-mediated expression.

FBXO23 targets FLCN for proteasomal degradation (A): A library of V5-tagged F-box O family plasmids were transfected in BEAS-2B cells and cell lysates were immunoblotted for FLCN, V5 or β-actin antibodies. (B): The densitometry data (FLCN/β-Actin) obtained from a are expressed as mean±SEM. (C): BEAS-2B cells were transfected with indicated amounts of V5-tagged FBXO23 or FBXO41 plasmid and cell lysates were immunoblotted for FLCN, V5, or β-actin antibodies. (D): The densitometry data (FLCN/β-Actin) obtained from (C) are expressed as mean±SEM. (E): V5-tagged FBXO23 or FBXO41 plasmid was transfected into BEAS-2B cells, and cell lysates were subjected to normal mouse IgG or V5 immunoprecipitation. The immunoprecipitates were immunoblotted for FLCN, V5 or GAPDH antibodies. Immunoblotting data are representative of three independent experiments. FLCN, folliculin.
We further confirmed that FBXO23 but not FBXO41 associates with FLCN by immunoprecipitation (figure 5E).
Phosphorylation at serine 62 promotes FLCN degradation in response to CS
Degron is the specific sequence within the substrate which are often post-translationally modified to affect their binding to E3 ligase.20 Multiple phosphorylation sites of FLCN have been reported and we were intrigued to study role of phosphorylation in CS induced FLCN degradation.8 We co-treated Beas-2B cells with CSE and several kinase specific inhibitors. Our data showed that cells with an ATM specific inhibitor Ku-55933 resistant to CSE induced FLCN degradation, suggesting that ATM may phosphorylate FLCN to facilitate its degradation on CSE treatment (figure 6A). We further confirmed that ATM siRNA stabilised FLCN protein and blocked the physical interaction between FLCN and FBXO23 compared with siCtrl in CSE-exposed cells (figure 6B and C). We then generated an alanine substitution at the major phosphorylation site S62 of FLCN and found this FLCN S62A mutant remained stable on CSE treatment compared with wild type (WT) control (figure 6D and E). Next, HA immunoprecipitation was performed to examine the relative expression of phosphorylated serine of the HA-tagged FLCN WT and FLCN S62A proteins in CSE-treated cells. S62A mutation attenuated phosphorylated serine relative to WT in CSE-treated cells (figure 6F). Furthermore, FLCN S62A decreased levels of CSE-induced cytotoxicity compared with FLCN WT (figure 6G). These data indicate that phosphorylation plays a key role in the ubiquitination and degradation of FLCN induced by CSE.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phosphorylation regulates FLCN degradation induced by the cigarette smoke (A): BEAS-2B cells were treated with 2% CSE together with Nu7441(10 µM), VE-821(10 µM) or KU-55933 (10 µM) for 24 hours. Cell lysates were immunoblotted with FLCN, phosphorylated ATM (pATM) or total ATM antibodies. (B): BEAS-2B cells were transfected with ATM siRNA (siATM) or scrambled siRNA (siCtrl) for 48 hours and further cultured with or without 2% of CSE for 8 hours. The cell lysates were immunoblotted for ATM, FLCN, or β-actin antibodies. (C): BEAS-2B cells were transfected with siATM or siCtrl for 24 hours, and cells were further transfected with FBXO23 plasmid and cultured another 48 hours. These cells were treated with 2% of CSE for 4 hours, and cell lysates were subjected to V5 immunoprecipitation. The immunoprecipitates were immunoblotted for V5, FLCN, GAPDH, or ATM, antibodies. Immunoblotting data are representative of three independent experiments. (D): HA-tagged FLCN (WT) or S62A mutant plasmid was transfected into BEAS-2B cells for 24 hours. The cells were treated with 2% CSE for up to 8 hours. Cell lysates were immunoblotted with HA or β-actin antibodies. (E): The densitometry data (HA/β-Actin) obtained from (D) are expressed as mean±SEM. (F): FLCN WT or FLCN S62A plasmid was transfected into BEAS-2B cells, and cell lysates were subjected to HA immunoprecipitation. The immunoprecipitates were immunoblotted for phosphoserine, HA, or GAPDH antibodies. Immunoblotting data are representative of three independent experiments. (G): BEAS-2B cells were transfected with FLCN WT or S62A and treated with 2% CSE overnight. Cell viability was measured by MTT assay. Data are expressed as a mean±SEM for three independent experiments with triplicate samples. ATM, ataxia‐telangiectasia mutated; CSE, cigarette smoke extract; FLCN, folliculin; HA, haemagglutinin; WT wild type.
Discussion
We demonstrate that (i) smokers with COPD exhibit a marked decrease in FLCN protein expression in human lungs compared with smokers without COPD; (ii) CS exposure decreases FLCN levels in human lung epithelial Beas-2B cells; (iii) FLCN protects against CSE-induced cell death; (iv) FLCN is regulated at the level of protein stability through ubiquitylation by the SCFFBXO23 complex; (v) phosphorylation at serine 62 (S62) in FLCN facilitates its degradation on CSE treatment. These data suggest that CS enhances the degradation of FLCN protein via the ubiquitin-proteasome mechanism and thereby augments epithelial cell death.
Cigarette smoking is a major risk factor for the development of emphysema and spontaneous pneumothorax. Likewise, the majority of BHD syndrome patients harbouring the loss-of-function mutations of FLCN develops pulmonary cysts and pneumothorax.21 We speculate that FLCN loss secondary to smoking may mimic the lung manifestations of BHD syndrome.
Cigarette smoking increases total cellular polyubiquitination level in lung epithelial cells.22 Our previous study revealed that CS exposure promoted ubiquitination mediates degradation of transforming acidic coiled-coil–containing protein 2, which also has a protective role in cell death and serves as a critical DNA repair factor.13 Similar to these findings, we confirmed that decreased FLCN protein levels in smokers with COPD and mice exposed to CS. These findings are important because we also found that FLCN depletion sensitises cells to CSE-induced cell death.
There is a growing interest in understanding how dysregulation of protein stability contributes to the pathogenesis of lung diseases.23 24 Our data indicates that FLCN is a labile protein with a half life of approximately 6 hours and that CSE can lead to FLCN degradation via the UPS. Among the FBXO family members, a subunit of the SCF E3 ligase, FBXO23 interacts with FLCN and FBXO23 overexpression reduces endogenous FLCN levels. FBXO23, also named tetraspanin 17, is a member of the tetraspanin superfamily which may be involved in the regulation of signalling mediated by adhesion, receptors like epidermal growth factor receptor and intracellular signalling.25 FBXO23 forms complexes with ADAM10 to maintain normal VE-Cadherin expression which is required for lymphocyte transmigration.26 A recent report also demonstrated a link between upregulation of FBXO23 with poor prognosis in patients with GBM (Glioblastoma multiforme) patients.27 However, the function of FBXO23 remains largely unknown.
The molecular mechanism of substrate recruitment by FBXO23 may help to better understand the biological function of this F-box protein. A degron is a short degradation motif within the substrates for binding to the F-box proteins. The canonical phosphodegron is the best-characterised molecular recognition signatures within substrates to facilitate F-box protein binding.28 Multiple phosphorylation sites within FLCN mediated through mTOR and 5' adenosine monophosphate-activated protein kinase (AMPK) regulates the binding of FLCN to FLCN-interacting protein 1 (FNIP1).29 30 S62 has been demonstrated as a major phosphorylation site of FLCN which regulates its binding to the AMPK.31 A recent study reported that ATM induced phosphorylation of AT-rich interactive domain-containing protein 1A to promote its degradation.32 Consistent with these results, we found that a specific ATM inhibitor treatment or ATM knock down can inhibit FLCN degradation induced by CSE, suggesting that ATM may phosphorylate FLCN to facilitate its degradation. Several phosphorylated forms of FLCN, present in cultured renal tumour cell lines, are regulated by mTOR and AMPK (5′ adenosine monophosphate-activated protein kinase).29 Although the specific amino acid sites are not identified, phosphorylation of FLCN increases the physical interaction with FNIP1 and may regulate the protein stability of FLCN. These are very interesting data, but there are 58 serine and 26 threonine residues in a total of 579 amino acids of FLCN. Future studies are necessary to identify specific phosphorylation site(s) that regulate protein stability in an unstressed culture condition. The mutant of S62A in FLCN is resistant to degradation, suggesting that S62 is a potential phosphosite for recognition by FBXO23. Further studies on the regulatory mechanism of FLCN phosphorylation are important to examine this relationship.
The purpose of this study is to investigate another detrimental effect of smoking, loss of FLCN, a tumour suppressor. Future studies will be necessary to investigate a potential causal role of smoking-induced FLCN loss in the development of COPD using the loss-of-function and gain-of-function in vivo model. Further, we used the limited sample size used for statistical analysis for both human and animal studies.
In summary, our studies show that cigarette smoking is a secondary cause of FLCN loss via the UPS and thereby promotes epithelial cell death. Our findings suggest that a canonical phosphodegron is involved in the regulation of FLCN stability. Cigarette smoking may mimic the lung manifestations of BHD due to FLCN deficiency.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the Institutional Review Board of the University of Pittsburgh (#STUDY19110180). Participants gave informed consent to participate in the study before taking part.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors XL, YJ and TN conceived and designed the experiments; XL, ZL and YL performed the experiments; KT, HS, CZ and TN analysed the data; YL, JCS, MR, MMM, YJ and TN contributed reagents/materials/analysis. TN is the guarantor, accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding This manuscript was supported, in part, by the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development by a Merit Review award CX001048 and CX000105 (TN). This work was also supported by American Heart Association (TN); NIH grants HL149719 (TN) and GM132127 (YJ). Frozen lung parenchymal tissue samples from smokers without COPD and with severe COPD were provided by the tissue core of University of Pittsburgh and LTRC, respectively.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.