Article Text

Statistics from Altmetric.com
Secretion of mucus into the airway lumen is a fundamental host defense mechanism that enables trapping of pathogens and other inhaled particles and their subsequent removal from the airways via mucociliary escalator. The numbers of mucus-producing goblet cells are tightly controlled to secure the baseline mucin levels on the airway surface, while preventing mucus plugging of small bronchioles, where these cells are rare or absent.1 Maintaining a proper number of mucus-producing cells in the airways is a function of tissue-resident basal stem cells, which, under control of physiological or injury-associated signals, regenerate airway epithelium with a distinct proportion of basal, ciliated and secretory cells.
Increase in goblet cell numbers or mucous cell hyperplasia (MCH) occurs in response to pathogens, oxidants, toxins, particles and cigarette smoke, leading to a transient mucus hypersecretion that normally disappears after the stimuli are no longer present. In chronic lung diseases, such as asthma and chronic obstructive pulmonary disease (COPD), overproduction of mucus persists over time contributing to clinical symptoms. Long-term maintenance of MCH, a morphological basis of chronic mucus hypersecretion in these conditions, can result from sustained activation of airway basal cells or their progenies by disease-associated signals that promote their excessive differentiation toward mucus-producing cells.
Several pathways are known to promote MCH by modifying the fate of airway basal cells. Airway inflammation driven by T helper (Th)2 cells, characteristic for asthma, promotes MCH via interleukin (IL)-13 that shifts the fate of airway basal cell-derived progenitors to the goblet cell lineage by activating Notch signalling required for differentiation of airway basal cells into secretory cells.2 3 Th17-derived IL-17 associated with neutrophilic airway inflammation in severe asthma and COPD exacerbations can promote MCH via Notch2-dependent signalling in airway basal cells.3
Cigarette smoking, the major risk factor for COPD, can promote MCH independent of inflammation, by activating epidermal growth factor receptor (EGFR) signalling in airway basal cells.4 Chronic mucus hypersecretion, or chronic bronchitis, is common among smokers and associated with the development of COPD. Although smoking-induced MCH is reversible, in the small airways, the primary site of airway obstruction in COPD, MCH can persist after smoking cessation.5 In COPD patients, chronic mucus hypersecretion is associated with more frequent exacerbations and a more rapid decline in lung function.6
What mechanisms mediate sustained MCH in COPD airways? In Thorax, Jing et al 7 address this question by evaluating the responses of epithelia regenerated in vitro by airway basal cells, isolated from subjects with or without COPD to rhinovirus infection, the common cause of COPD exacerbations.8 Consistent with previous reports,9 airway basal cells from COPD patients generated the epithelium with a higher number of mucus-producing cells, suggesting that a ‘memory’ of MCH is maintained in these cells, even after separation from disease-associated in vivo microenvironment. Strikingly, the authors found that epithelia derived from COPD, but not normal airway basal cells responded to rhinovirus infection with further increase in goblet cell numbers, indicative of MCH. Rhinovirus-induced MCH was replicated in a murine in vivo model, which recapitulates several features of COPD airway disease, and persisted for several days after rhinovirus was no longer detectable in the airways. Thus, in addition to causing acute COPD exacerbations, rhinovirus infection may have a long-term impact on disease progression by promoting airway epithelial remodelling and chronic mucus hypersecretion, once the disease is established. Indeed, in an earlier study, experimental rhinovirus infection caused long-term respiratory symptoms and airway obstruction in COPD subjects, but not in individuals without airway disease.10
What makes the airway epithelium in COPD susceptible to rhinovirus-induced MCH? In COPD, the airway epithelium undergoes structural changes, which, apart from MCH, include basal cell hyperplasia, characterised by that is, increased number of basal cells and basal-like undifferentiated cells, and squamous metaplasia, with the appearance of squamous cells that replace ciliated cells. These lesions are accompanied by loss of tight junctions that control the permeability of the epithelial barrier. When the airway epithelium acquires this aberrant pattern, it becomes ‘an easy prey’ for rhinovirus that preferentially targets undifferentiated basal cells, which express a rhinovirus receptor, intercellular adhesion molecule 1, or cells undergoing squamous differentiation.11 12
Once rhinovirus gets an access to basal cells or basal cell-derived undifferentiated cells, it suppresses junctional barrier formation and ciliated cell differentiation by inhibiting mechanisms necessary for the establishment of epithelial polarity, while promoting the generation of mucus-producing cells.13 Rhinovirus exerts this effect via an EGFR-dependent mechanism, and this occurs when the airway epithelium is not properly differentiated.13 A similar phenotype is induced by smoking-associated EGFR signalling in airway basal cells.4 It is, therefore, likely that the pathological process in COPD driven by smoking may produce a chronic injury-like airway epithelial phenotype, particularly susceptible to rhinovirus infection.
What mechanism underlies rhinovirus-induced MCH in COPD airway epithelium? To address this question, Jing et al 7 performed transcriptome analysis, which identified two receptors of Notch pathway, Notch1 and Notch3, and the downstream effector Hey1, as being upregulated in rhinovirus-infected epithelia derived from COPD airway basal cells, but not those from the normal airways. Upregulation of Notch3 and Hey1 was also observed following rhinovirus infection in the mouse COPD airway model. Pharmacological inhibition of Notch signalling reduced rhinovirus-induced MCH in both models. This effect was reproduced when Notch3 was selectively knocked-down in COPD airway epithelial cells, accompanied by downregulation of FOXA3, a transcription factor implicated in airway MCH in response to rhinovirus.14 Rhinovirus-induced MCH in COPD airway epithelia was independent of EGFR, which mediates the effect of rhinovirus in non-COPD airway epithelial cells,13 and IL-13, an MCH-promoting cytokine elevated during Th2-driven inflammation.3 These data point towards a novel, Notch3-dependent epithelial-autonomous mechanism that mediates rhinovirus-induced airway MCH in COPD.
Notch signalling, initiated by activation of cell-surface Notch receptors by transmembrane ligands Delta-like and Jagged on neighbouring cells, plays a key role in mediating secretory cell differentiation in the airway epithelium.2 It has been shown that Notch3 marks airway basal cell-derived undifferentiated progenitors and that these cells are maintained by Jagged expressed by adjacent basal cells, preparing these progenitors for differentiation into secretory cells.15 The role of the Notch pathway in COPD has been controversial because MCH occurs in this disease despite the broad smoking-dependent downregulation of Notch pathway components in the airway epithelium.16 Whereas the latter was found in COPD subjects in the exacerbation-free period, Notch3-dependent MCH was observed by Jing et al 7 in COPD airway epithelium after rhinovirus infection, particularly relevant to COPD exacerbations.
When we link findings of Jing et al 7 to existing knowledge about COPD pathogenesis, a novel mechanistic model emerges, which explains the development of persistent MCH in COPD as a gradually progressing process that includes three events (figure 1). The first event, smoking-associated airway epithelial remodelling, develops due to EGFR-dependent reversible reprogramming of airway basal cells and leads to the acquisition of an aberrant differentiation pattern susceptible to rhinovirus. The second event, likely occurring during COPD exacerbations, is driven by rhinovirus infection, which promotes persistent MCH by activating Notch3-Hey1-FOXA3 axis in basal cell-derived progenitors. The third, most mysterious, event is characterised by stable reprogramming of basal cells enabling these cells to produce MCH independently of smoking or infection. In COPD airways, these events may occur simultaneously leading to chronic mucus hypersecretion.

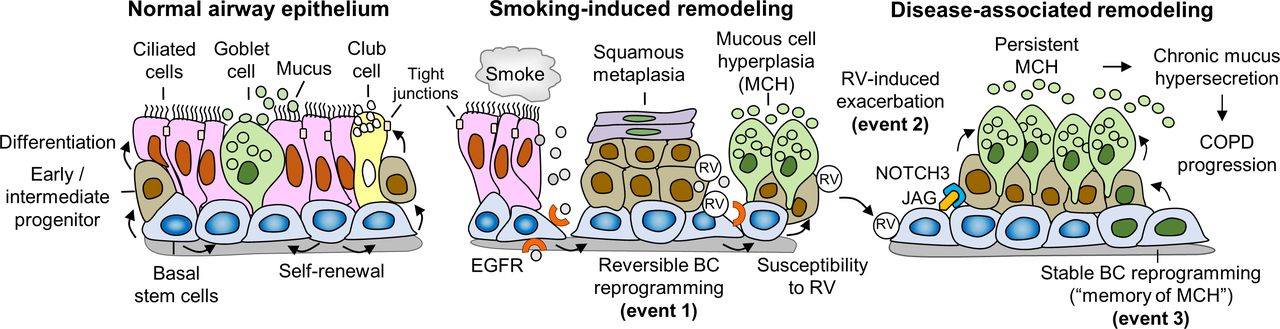{kind=link}
A three-event model of airway MCH pathogenesis in COPD. (Left panel) The normal airway epithelium is maintained by basal stem cells (BCs)capable of self-renewal and differentiating into ciliated cells and secretory cells, including mucus-producing goblet cells and non-mucous secretory club cells. This process involves generation of early/intermediate progenitors, or para-BCs, which represent precursors of differentiated cell populations, and formation of tight junctions between differentiated cells that control epithelial barrier permeability. (Middle panel) Smoking causes reversible histological lesions in the airway epithelium (first event), including BC hyperplasia, squamous metaplasia, MCH and loss of junctional barrier integrity, by inducing exaggerated EGFR signalling in BCs. Acquisition of this aberrant differentiation pattern makes the airway epithelium susceptible to RV infection, which further promotes airway remodelling phenotypes in the injured and repairing airway epithelia. (Right panel) In COPD, RV infection, which causes COPD exacerbations, and, as reported by Jing et al,7 promotes persistent MCH by activating Notch3-dependent signalling (second event), possibly in undifferentiated para-BCs by the Notch ligand JAG expressed in adjacent BCs.15 Stable, disease-specific reprogramming of BCs in COPD airways, likely through epigenetic alterations (third event), renders these cells capable of continuously producing MCH independent of smoking or RV infection (‘memory’ of MCH). These three events may occur simultaneously and lead to chronic mucus hypersecretion, contributing to symptoms and disease progression. BC, basal stem cell; COPD, chronic obstructive pulmonary disease; EGFR, epidermal growth factor receptor; MCH, mucous cell hyperplasia; RV, rhinovirus
An important aspect of rhinovirus infection in COPD is that it often leads to secondary bacterial infection,8 which may develop due to altered mucus clearance and further sustain MCH. For example, Haemophilus influenzae, a bacterial pathogen commonly colonising the airways during COPD exacerbations, causes inflammation with increased levels of IL-17,17 a cytokine that stimulates MCH.3 Thus, rhinovirus infection in COPD represents an example for an altered disease tolerance principle in pathophysiology,18 in which inability to tolerate host response to a pathogen, rather than pathogen itself, becomes a driver of disease pathogenesis.
A three-event model of MCH pathogenesis in COPD outlined in figure 1 implies that different therapies might be effective at different biological phases of the disease. Restoration of epithelial differentiation through smoking cessation and inhibition of smoking-associated signalling pathways, such as those mediated by EGFR, could be beneficial in preventing rhinovirus infection. During rhinovirus-induced exacerbations, antiviral drugs and modulators of biological pathways, employed by rhinovirus to cause mucociliary dysfunction, would be the therapies of choice. Pharmacological inhibition of Notch signalling could be particularly valuable in this regard, since, in addition to reducing MCH, it reciprocally restores ciliated cell differentiation,19 which is suppressed when Notch pathway is activated.20 Selective targeting Notch3 signalling is even more attractive, since it may reduce rhinovirus-induced MCH, as demonstrated by Jing et al,7 without inactivating other Notch receptors necessary for maintaining other secretory lineages, such as club cells, whose numbers decrease when MCH develops.1
Perhaps, the most intriguing question is how to erase the ‘memory’ of susceptibility to MCH, which is maintained in COPD airway basal cells, possibly through disease-specific epigenetic modifications. The answer to this question requires further investigation into the nature of long-term molecular changes in airway basal cells leading to progressive airway remodelling in this disease.
Acknowledgments
Work in the Author’s laboratory is supported by National Institutes of Health (grants R01HL123544 and R01HL127393).
References
Footnotes
Contributors RS is the sole contributor.
Funding This study was funded by National Heart, Lung, and Blood Institute (R01HL123544, R01HL127393).
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- Chronic obstructive pulmonary disease